BAND
Periodic DFT for nanotubes, surfaces, and bulk
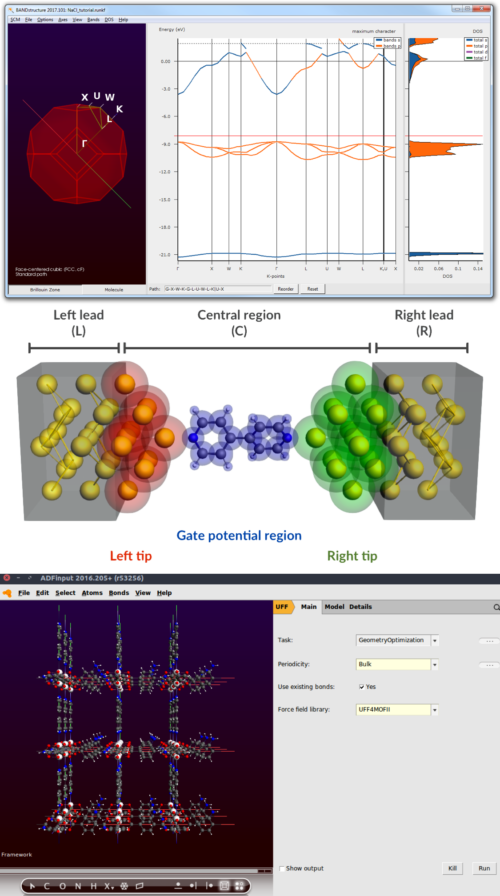
BAND, the accurate periodic density functional theory (DFT) code of the Amsterdam Modeling Suite shares many powerful features with our molecular DFT code ADF. Using atomic orbitals for periodic DFT calculations has many advantages over plane waves like a proper treatment of surfaces, efficient computations of sparse matter, and more direct and detailed analysis methods. For fast calculations on dense systems, we also ship the plane wave code Quantum ESPRESSO.
Efficient & Accurate Periodic DFT with BAND and QM/MM
SCM’s expert Pier Philipsen demonstrates useful techniques and tricks to obtain accurate DFT-quality results for large periodic models.