Previous EU Projects
See also our current EU projects.
ReaxPro
SCM is proud to have lead the ambitious ReaxPro project to integrate atomistic, mesoscale, and macroscale modeling tools in a comprehensive multi-scale reactor modeling platform to aid BASF, Shell, DowDuPont & Johnson Matthey to optimize their catalytic reactors.
AutoReactPro
SCM and Hafnium Labs collaborated to develop software to automatically predict side reactions to help design chemical processes more safely and efficiently.
AutoCheMo
Automatic generation of Chemical Models is a joint research and training project (European Industrial Doctorate) coordinated by SCM, with Ghent University and RWTH Aachen University. AutoCheMo will develop new and improved tools for modeling chemical processes in industrial reactors.
S4CE
The Science4CleanEnergy multi-disciplinary consortium which will further advance carbon-capture storage technology. At SCM we will focus on further developing ReaxFF -including acceleration and parametrization- to model such complex processes as CO2 storage in clay materials.
MaLeR
Machine Learning applied to Reactivity. Marie Curie fellow Matti Hellström will improve the accuracy and automated ReaxFF parametrization with machine learning.
PolySolv
SME innovation associate Nick Austin has implemented methods to broaden the scope of COSMO-RS to deal with larger systems, in particular polymers.
NET
Tomáš Trnka worked as an SME innovation associate methods to accelerate ReaxFF for accessing longer timescales for complex, chemical reaction dynamics.
TCCM
As an associate partner, SCM hosted secondments of students in the Theoretical Chemistry and Computational Modeling network, as well as participating in hands-on workshops. The largest project we are involved in is on organic electronics, in collaboration with Ria Broer’s group in Groningen.
DEFNET
New methods for efficiently and accurately treating DEFects in NETwork materials (MOFs) were developed by SCM and other partners (Ghent, Bochum) in this European Training Network. Together with experimental groups and industrial partners the aim is to engineer materials for their catalytic and gas sorption properties. At SCM we are working on improving ReaxFF force fields and making ReaxFF parametrization tools.
MoWSeS
The EPFL-led ITN MoWSeS (http://mowses.epfl.ch) focuses on nanoelectronics with two-dimensional dichalcogenides. In this project SCM and Jacobs are DFT(B) method developing partners, in synergy with the QUASINANO project, for heat transport, charge transport and optical properties of two-dimensional semiconductors. This has already resulted in the successful implementation of NEGF in DFTB for calculating transport properties of large MoWSeS structures such as rippled nanosheets and multi-walled nanotubes.
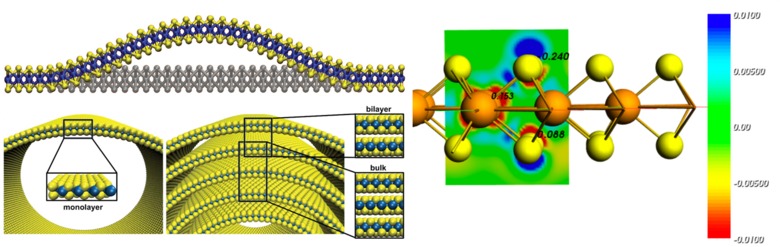
Charge transport properties of MoS depend strongly on deformations (top left, Adv. Mater. 25, 5473 (2013)) and strain (bottom left, Scientific Reports 3, 2961 (2013)). The band gap of 2D semiconductors decreases with increasing electric fields (right, Phys. Chem. Chem. Phys. 16, 11251 (2014)).
PROPAGATE
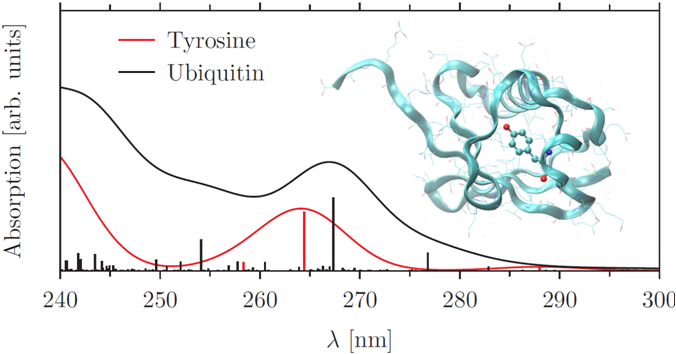
Three European Industrial Doctorate candidates develop QM/MM methods and ab initio molecular dynamics for ground and excited states in the PROPAGATE project, with the SCM and Jacobs University (now: Leipzig) as partners and the VU as an associate partner. In this project, UV/VIS spectra with TDDFTB have been implemented as well as density matrix purification for faster, linear-scaling DFTB calculations. More recently, excited state gradients have been implemented for TDDFTB, so that one can optimize excited states or calculate vibrational progression of excited states. Time-dependent DFTB enables calculation of UV/VIS spectra of entire proteins. The TD-DFT+TB approach uses similar approximations as TDDFTB but using DFT-based orbitals, which enormously speeds up calculation time with respect to TDDFT. R. Rüger, E. van Lenthe, T. Heine, L. Visscher, Tight-Binding Approximations to Time-Dependent Density Functional Theory – a fast approach for the calculation of electronically excited states J. Chem. Phys. 144, 184103 (2016)
Fortissimo
SCM participates in an experiment in Fortissimo (www.fortissimo-project.eu), which aims to bring cloud-based HPC solutions to small and medium enterprises (SMEs). The ADF and ReaxFF codes are being accelerated for hybrid CPU-GPU architectures and we are also setting up ADF for remote (cloud) computing with web-based visualization. We are proud to offer ADF in the cloud through our partner CrunchYard.
QUAntum-mechanical SImulations for the NANOscale
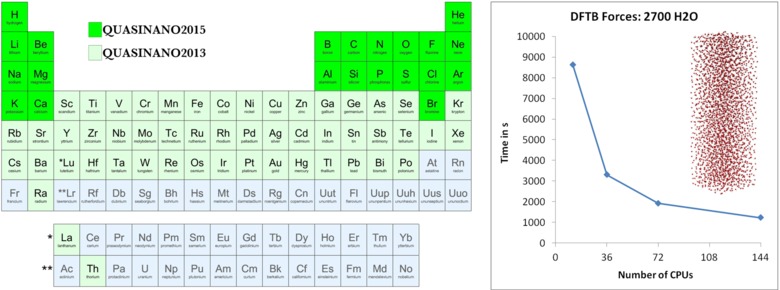
SCM’s first succesful EU project was in collaboration with the Heine group of Jacobs University in Bremen: QUASINANO. The project runs from October 2010 to September 2014. Density-functional based tight-binding (DFTB) methods were developed to achieve simulations at the nanoscale (length and time). Through various secondments the DFTB code has been optimized and its functionality considerably expanded (MD, property visualization, TDDFTB, parameters). Left: DFTB Parameters for the Periodic Table Part 1: Electronic Structure (J. Chem. Theory Comput. 9, 4006 (2013)); Part 2: Gradients from H-Ca (J. Chem. Theory Comput.11, ASAP (2015)). Right: parallel scaling of DFTB forces for a box with 2700 H2O molecules. We also have very interesting local collaborative projects.