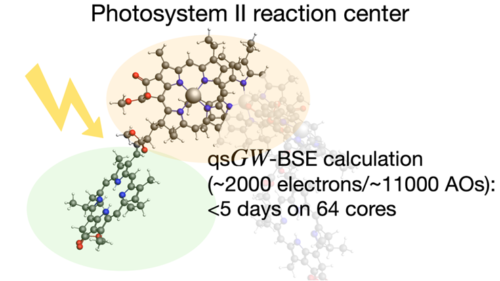
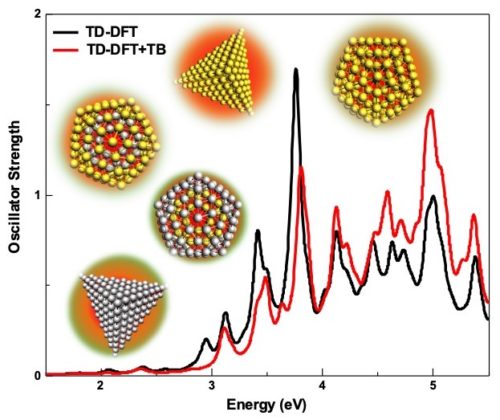
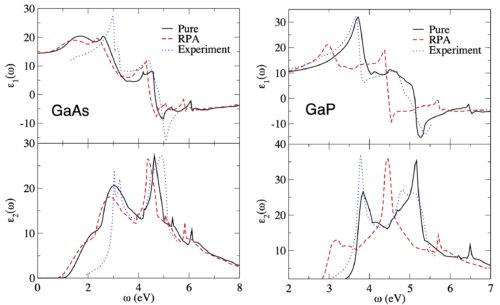
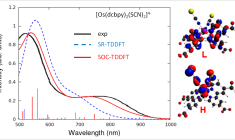
Calculate UV/VIS spectra
Extensive time-dependent density functional theory (TDDFT) capabilities in ADF ensures the efficient and accurate modeling of UV/VIS spectra, phosphorescent processes and excited state geometries or frequencies for simple and complex molecules. Easily calculate, visualize and analyze UV/VIS spectra with TDDFT and fast approximations with our graphical user interface.