Properties¶
Here, we illustrate the required inputs for calculating different properties. Refer to the governing equation at calculation of properties.
Vapor pressure¶
The vapor pressure of a mixture can be calculated with:
PROPERTY vaporpressure
End
In case of a mixture the mole fraction of each compound of the solvent should be given with the subkey FRAC1 of the key COMPOUND for each compound. In case of a mixture also activity coefficients, and excess energies are calculated.
To calculate pure compound vapor pressures for more than one compound use:
PROPERTY purevaporpressure
End
It is possible to calculate the vapor pressure for a temperature range, see key TEMPERATURE.
The input pure compound vapor pressure will be used in the calculation of the vapor pressure of this compound if it is supplied with the key COMPOUND for this compound. If it is not specified then it will be approximated using the COSMO-RS method.
Boiling point¶
The boiling point of a mixture can be calculated with the block key:
PROPERTY boilingpoint
End
In case of a mixture the mole fraction of each compound of the solvent should be given with the subkey FRAC1 of the key COMPOUND for each compound. In case of a mixture also activity coefficients, and excess energies are calculated.
To calculate pure compound boiling points for more than one compound use:
PROPERTY pureboilingpoint
End
It is possible to calculate the boiling temperature for a pressure range, see key PRESSURE.
The input pure compound vapor pressure will be used in the calculation of the vapor pressure of this compound in the mixture if it is supplied with the key COMPOUND for this compound. If it is not specified then it will be approximated using the COSMO-RS method.
The COSMO-RS calculation of the boiling temperature of a solvent is performed with an iterative method. The temperature is varied until the calculated vapor pressure is within a certain threshold of the desired pressure.
Links COSMO-RS GUI tutorial: boiling point of a solvent [1]
Solvent flash point¶
The flash point (lower flammable limit) of a compound is the lowest temperature at which the vapor of the compound forms an ignitable mixture in air. The COSMO-RS module can calculate the flash point of a mixture. The COSMO-RS module, however, does not calculate or predict the flash point of pure compounds. The COSMO-RS method is used to calculate the partial vapor pressures of each compound in the mixture, and it uses Le Chatelier’s mixing rule to calculate the flash point of this mixture in the gas phase. Input pure compound flash points should be provided by the user, with the subkey FLASHPOINT flashpoint of the key COMPOUND.
PROPERTY flashpoint
End
The mole fraction of each compound of the solvent should be given with the subkey FRAC1 of the key COMPOUND for each compound.
Partition coefficients (LogP)¶
The partition coefficient of a compound in a mixture of two immiscible solvents, can be calculated with:
PROPERTY logp
{VOLUMEQUOTIENT volumequotient}
End
volumequotientIf the subkey VOLUMEQUOTIENT is included the volumequotient will be used for quotient of the molar volumes of solvent 1 and solvent 2 instead of calculated values.
The mole fraction of each compound of the solvent 1 and solvent 2 should be given with the subkey FRAC1 and subkey FRAC2 of the key COMPOUND for each compound, respectively. In case of partly miscible liquids, like, for example, the Octanol-rich phase of Octanol and Water, both components have nonzero mole fractions. The compounds that are included without a given mole fraction are considered to be infinite diluted solutes. The partition coefficients are calculated for all compounds.
One can use some compounds that are present in $AMSHOME/atomicdata/ADFCRS (Water, 1-Octanol, Benzene, Ethoxyethane, Hexane), or one can use compounds from the ADFCRS-2010 database. For example, for Octanol/Water partition coefficients one can use:
Property logp
VolumeQuotient 4.93
End
Compound "$AMSHOME/atomicdata/ADFCRS/1-Octanol.coskf"
frac1 0.725
End
Compound "$AMSHOME/atomicdata/ADFCRS/Water.coskf"
frac1 0.275
frac2 1.0
End
Links COSMO-RS GUI tutorial: partition coefficients (log P) [1, 2], Octanol-Water partition coefficients (log POW ) [1]
Activity coefficients solvent and solute¶
The mole fraction of each compound of the solvent should be given with the subkey FRAC1 of the key COMPOUND for each compound. The compounds that are included without a given mole fraction are considered to be infinite diluted solutes. The activity coefficients are calculated for all compounds.
PROPERTY activitycoef
{DENSITYSOLVENT densitysolvent}
End
densitysolventIf the subkey DENSITYSOLVENT is included the densitysolvent will be used for the density of the solvent (kg/L) instead of calculated values. Relevant for the calculation of the Henry’s law constant.
The input pure compound vapor pressure will be used in the calculation of the partial vapor pressure of this compound in the mixture if it is supplied with the key COMPOUND for this compound. If it is not specified then it will be approximated using the COSMO-RS method. Relevant for the calculation of the Henry’s law constant.
The Henry’s law constants are calculated in 2 units. The Henry’s law constant kH is the ratio between the liquid phase concentration of a compound and its partial vapor pressure in the gas phase. The dimensionless Henry’s law constant kH cc is the ratio between the liquid phase concentration of a compound and its gas phase concentration.
Also calculated is \(\Delta G_{solv}^{liq-solv}\) , which is the solvation Gibbs free energy from the pure compound liquid phase to the solvated phase, and \(\Delta G_{solv}^{gas-solv}\) , which is the solvation Gibbs free energy from the pure compound gas phase to the solvated phase, with a reference state of 1 mol/L in both phases. In addition a Gibbs free energy is calculated which is the free energy of the solvated compound with respect to the gas phase energy of the spin restricted spherical averaged neutral atoms, the compound consist of. Note that zero-point vibrational energies are not taken into account in the calculation of this free energy. This energy could be used in the calculation of \(pK_a\) values.
Links COSMO-RS GUI tutorial: activity coefficients [1, 2], solvation free energies [1], Henry’s law constants [1], \(pK_a\) values [1]
Solubility¶
The solubility of solutes in 1 mixture can be calculated with:
PROPERTY solubility
End
The mole fraction of each compound of the solvent should be given with the subkey FRAC1 of the key COMPOUND for each compound, and should add up to 1.0. The solutes should have zero molar fraction in the solvent. The solubility of 1 solute in pure solvents can be calculated with
PROPERTY puresolubility
End
The mole fraction of each pure solvent should be 1.0, and should be set the subkey FRAC1 of the key COMPOUND for each compound. The solute should have zero molar fraction in the solvent. It is possible to calculate the solubility of a solute at a temperature range, see key TEMPERATURE.
For solubility calculations of a solid compound one should add the pure compound melting point \(T_m\) , pure compound enthalpy of fusion \(\Delta H_{fus}\) , and optionally the pure compound heat capacity of fusion \(\Delta C_p\) using the subkeys meltingpoint, hfusion, and cpfusion, respectively, of the key COMPOUND for this compound. The COSMO-RS method does not predict these \(\Delta H_{fus}\), \(\Delta C_p\), or \(T_m\) .
The assumption made in the solubility calculation may be invalid in case of a solubility of a liquid in a solvent, especially if the solubility of the solvent in the liquid is high. For binary systems one may check this by calculating the miscibility gap in the binary mixture of the two liquids. It is possible to calculate the solubility of a gas in a solvent, if one adds the subkey isobar and adds the partial vapor pressure partialvaporpressure (bar) of the gas as argument for the key PRESSURE:
PROPERTY solubility
isobar
End
PRESSURE partialvaporpressure
The solubility of a gas in a solvent can also be calculated using Henry’s law, which is valid for ideal dilute solutions, see see the key PROPERTY activitycoef. The COSMO-RS calculation of the solubility of a compound is performed with an iterative method, since the activity coefficient of the compound depends on the molar fraction of this compound.
Phase Stability Test¶
The Michelsen Tangent Plane Distance (TPD) stability test can be calculated with:
PROPERTY stability
End
COMPOUND "Water.coskf"
frac1 0.5
End
COMPOUND "1-Octanol.coskf"
frac1 0.5
End
The feed composition is given with the subkey FRAC1 of the key COMPOUND for each compound. The values of FRAC1 should sum to 1.0 over all compounds.
Advanced algorithm settings¶
The Michelsen TPD stability test [1] determines phase stability by searching for stationary points of the tangent-plane distance function \(TPD(w)\) in composition space. If a stationary point is found such that \(TPD(w^*) < 0\), the mixture is thermodynamically unstable and phase splitting is detected.
The governing equations and the algorithmic workflow are summarized in the flowchart below.
Algorithm outline
Governing equations
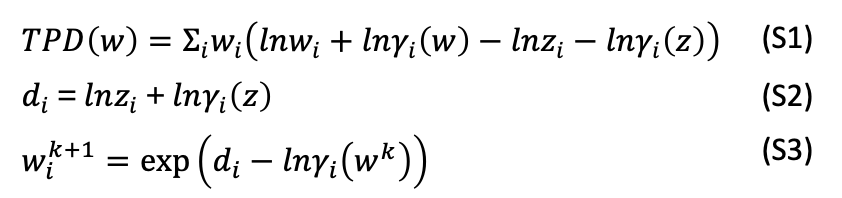
The TPD function (Eq. S1) is evaluated for trial compositions \(w\) relative to the feed composition \(z\). For convenience, \(d_i\) is defined as in Eq. S2.
The stationary condition is solved by a fixed-point iteration (Eq. S3), followed by normalization (\(\sum_i w_i = 1\)) and optional damping with factor \(\lambda\) as shown in the flowchart below.
Flowchart
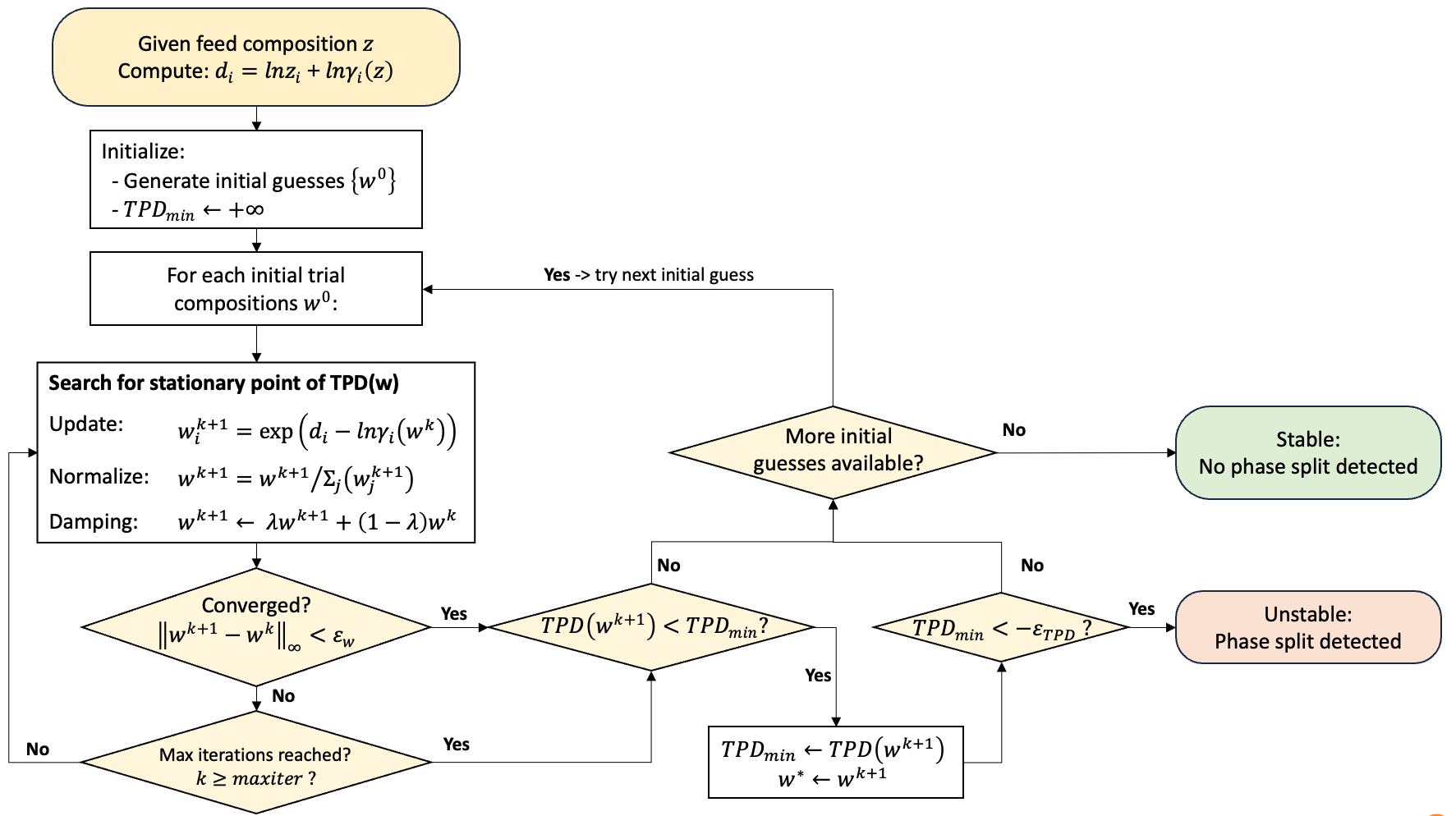
Given a feed composition \(z\), the algorithm first evaluates \(d_i\) using Eq. S2, then generates a set of initial trial compositions \(\{w^0\}\) and initializes \(TPD_{\min}=+\infty\).
For each initial guess \(w^0\), it searches for a stationary point of \(TPD(w)\) using the update-normalize-damp cycle shown in the flowchart, until either convergence
or the iteration limit is reached (\(k \ge \mathrm{maxiter}\)).
After each trial, if a lower value is found, the best-so-far minimum is updated:
After all initial guesses are processed, the mixture is classified as unstable if
Otherwise, it is classified as stable (no phase split detected).
Technical Parameters
TPD solver parameters
eps_tpdInstability tolerance \(\varepsilon\). The mixture is reported as unstable if \(TPD(w^*) < -\varepsilon\). (default:
1e-5)eps_wConvergence tolerance \(\varepsilon_w\) for the fixed-point iteration: stop when \(\lVert w^{k+1} - w^k \rVert_\infty < \varepsilon_w\). (default:
1e-8)maxiterMaximum number of iterations per initial trial composition. (default:
500)lamDamping factor \(\lambda\) used in the relaxed update of \(w\). (default:
0.7)xmin/gamminLower bound for compositions/activity coefficients to avoid log(0), division by zero, and numerical instability; values below this threshold are clamped and renormalized. (default:
xmin = 1e-8,gammin = 1e-8)
Optional initial guess
tpd_wUser-supplied trial composition \(w_i\) of component i for the Michelsen TPD stability test (molar or mass fraction). (default: 0.0, meaning no user-supplied initial guess)
If tpd_w values are provided for all compounds, they should represent a valid composition (sum to 1.0).
If no tpd_w values are provided, a built-in default set of trial-phase compositions is used.
Example¶
PROPERTY stability
End
COMPOUND "Water.coskf"
frac1 0.5
tpd_w 0.1
End
COMPOUND "1-Octanol.coskf"
frac1 0.5
tpd_w 0.9
End
TECHNICAL
TPD
eps_tpd 1e-5
eps_w 1e-8
maxiter 200
lam 0.7
END
END
Links COSMO-RS GUI tutorial: phase stability test [1]
Liquid-Liquid Equilibrium (LLE)¶
The liquid-liquid equilibrium (LLE) of a multicomponent mixture can be calculated with:
PROPERTY lle
End
COMPOUND "Water.coskf"
frac1 0.5
End
COMPOUND "1-Octanol.coskf"
frac1 0.5
End
The feed composition is given with the subkey FRAC1 of the key COMPOUND for each compound. The values of FRAC1 should sum to 1.0 over all compounds.
Optional initial guess
The initial guesses for the phase split composition can be supplied:
lle_xI0Initial guess for phase-I composition \(x_i^I\) for component i (molar or mass fraction). (default: 0.0 [no user-supplied initial guess])
If no initial guesses are provided, a stability test is used to generate a starting split. If the feed is stable (single phase), the calculation reports “no phase split detected”.
Note
The current implementation solves only two-phase liquid–liquid equilibrium (LLE).
For systems exhibiting three-liquid-phase equilibrium (LLLE), the solver does not perform a global multi-phase stability analysis. While a two-phase solution may still be obtained, the presence of an additional liquid phase cannot be automatically identified by the current algorithm.
Advanced algorithm settings¶
The LLE solver iterates on the distribution coefficients \(K\) to satisfy the isoactivity condition [1]. The governing equations and the algorithmic workflow are summarized in the flowchart below.
Algorithm outline
Governing equations

The solver uses the convention in Eqs. L1-L2, and solves the Rachford–Rice equation \(F(L)=0\) at fixed \(K\) (Eq. L3) to obtain phase compositions \(x^I\) and \(x^{II}\) (Eqs. L4–L5). The outer iteration updates \(lnK\) using the isoactivity residual \(g\) (Eq. L6) via the step-controlled update in Eq. L7.
Flowchart
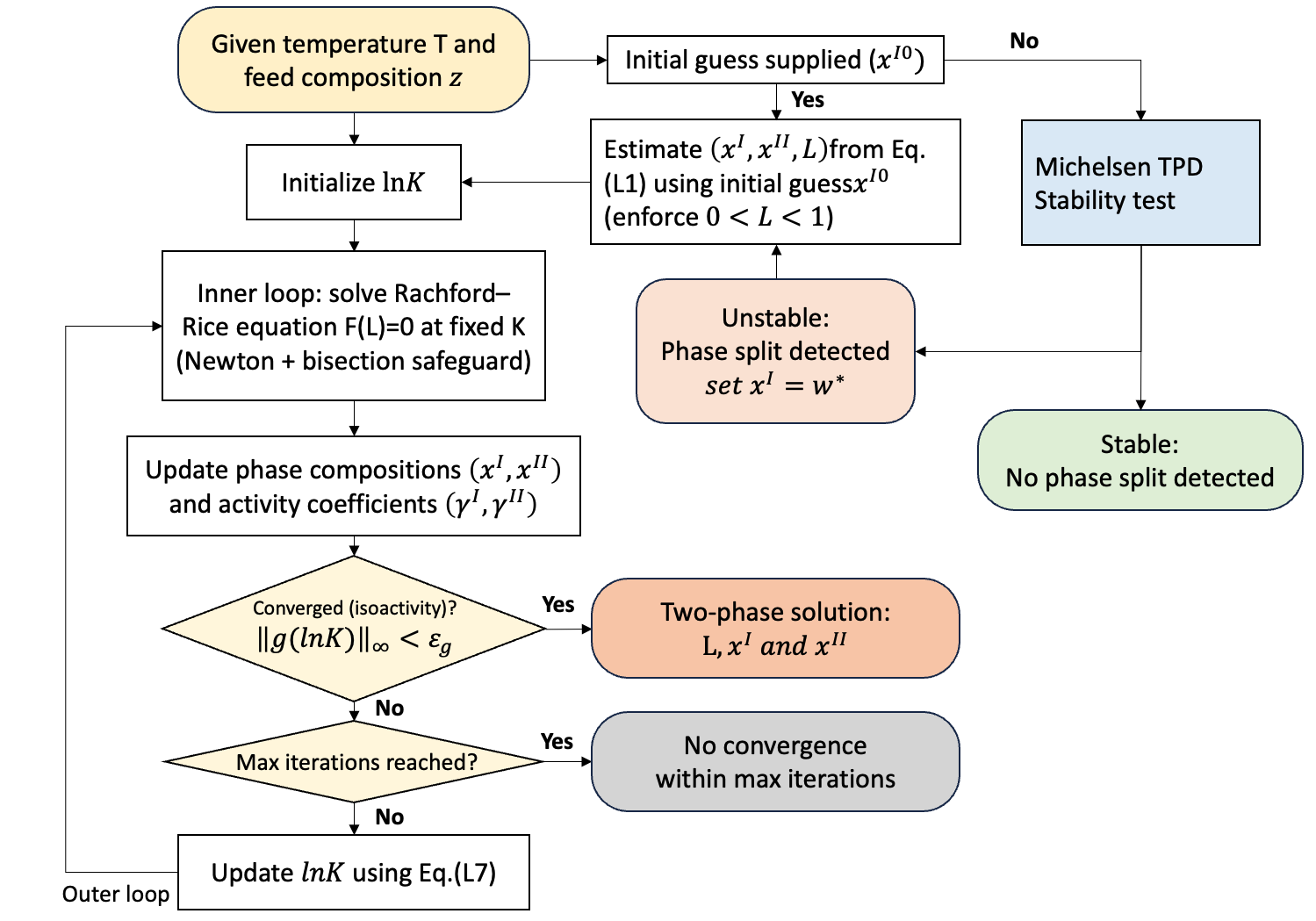
An initial guess can be supplied by the user via lle_xI0. If no guess is provided, \(w^{\min}\) is obtained from the TPD stability test and used as the initial phase-split composition.
At each outer iteration, convergence is checked using the log-isoactivity residual \(g_i = \ln(x_i^{II} \gamma_i^{II}) - \ln(x_i^{I} \gamma_i^{I})\) (Eq. L6). The solver reports convergence when \(\max_i |g_i| < \varepsilon_{g}\).
Technical Parameters
Outer-iteration convergence
eps_gConvergence tolerance \(\varepsilon_{g}\) on the log-isoactivity residual (Eq. L6): the solver checks \(\max_i |g_i|\). (default:
1e-6)maxiterMaximum number of outer LLE iterations (lnK updates). (default:
2000)
Step-size control for the lnK update
lnK_step_schemeStep-size scheme for the outer update \(\ln K \leftarrow \ln K - \lambda g\). Default:
3.1: fixed step (usestep_lnK)2: BB-short (Barzilai–Borwein short step)3: Mehra Algorithm III (adaptive; see Ref. [2])
step_lnKStep size \(\lambda\) for scheme
1. For other schemes it is used as the initial value. (default:1.0)dlnK_maxLimiter on the maximum \(\ln K\) change per iteration. The limiter follows Mehra et al. (1983, Eq. 40) [2]. Default:
6.0.\[\max_i |\Delta \ln K_i| \le dlnK_{\max}\]use_step_boundsEnable clipping of the step size \(\lambda\) to the interval \([\mathrm{step\_min}, \mathrm{step\_max}]\). Default:
false.step_min/step_maxBounds for the step size \(\lambda\) (clipping) to avoid stagnation (too small) and overly aggressive updates (too large). (default:
step_min = 0.002,step_max = 500.0)
Inner solve for phase fraction L (Rachford–Rice)
tolFResidual tolerance for the Rachford–Rice equation \(F(L)\). Convergence is accepted when \(|F(L)| < \mathrm{tolF}\). (default:
1e-8)tolLTolerance for phase-fraction \(L\) uncertainty (root-bracketing interval width); convergence is accepted when it is below
tolL. (default:1e-6)itLmaxMaximum number of L root‑finding iterations per LLE step. (default:
200)LminMinimum allowed phase fraction (physical/model bound). (default:
1e-6)
Robustness bounds
xminNumerical floor for LLE phase mole fractions (used in clipping, normalization, and log terms). (default:
1e-8)gamminNumerical floor for activity coefficients used in log terms. (default:
1e-8)marginNumerical safety margin in the L solver to keep denominators positive. (default:
1e-12)Lnum_minNumerical lower bound for phase-fraction safety; protects against an overly small Lmin. (default:
1e-12)
Notes¶
lnK_step_scheme = 3often improves convergence speed, but it may still fail for some cases.If the outer iteration fails to converge, first reduce
dlnK_max(and, if needed, increasemaxiter), then try a differentlnK_step_scheme.If the inner L-solver fails to converge, try increasing
itLmaxor relaxingtolF/tolL.
Example¶
PROPERTY lle
End
COMPOUND "Water.coskf"
frac1 0.5
lle_xI0 0.275
End
COMPOUND "1-Octanol.coskf"
frac1 0.5
lle_xI0 0.725
End
TECHNICAL
LLE
eps_g 1e-6
maxiter 2000
lnK_step_scheme 3
step_lnK 1.0
dlnK_max 6.0
tolF 1e-8
itLmax 200
END
END
lle_xI0 is an optional initial guess.
Links COSMO-RS GUI tutorial: liquid-liquid equilibrium [1, 2]
Binary mixture (VLE/LLE)¶
The COSMO-RS module can automatically calculate properties of a binary mixture, by calculating these properties for a number of different compositions.
Exactly two compounds should be given in the input file.
PROPERTY binmixcoef
{Nfrac nfrac}
{isotherm | isobar | flashpoint}
End
nfracNumber of different mixtures for which the binary mixture is calculated will be nfrac+5. Default value for nfrac is 10, which means 15 different mixtures.
isotherm | isobar |flashpointIf the subkey isotherm is included (default) the binary mixture will be calculated at a fixed temperature. If the subkey isobar is included the binary mixture will be calculated at a fixed vapor pressure. If the subkey flashpoint is included the flash point of the binary mixture will be calculated.
The input pure compound vapor pressure will be used in the calculation of the partial vapor pressure of this compound in the mixture if it is supplied with the key COMPOUND for this compound. If it is not specified then it will be approximated using the COSMO-RS method.
In case of a miscibility gap (LLE) data of the 2 immiscible liquid phases will be calculated. Also information about possible azeotropes will be calculated. With the COSMO-RS GUI, activity coefficients, (partial) vapor pressures, and excess energies can be viewed.
Links COSMO-RS GUI tutorial: vapor-liquid diagram binary mixture (VLE/LLE) [1, 2]
Ternary mixture (VLE/LLE)¶
The COSMO-RS module can automatically calculate properties of a ternary mixture, by calculating these properties for a number of different compositions. Exactly three compounds should be given in the input file.
PROPERTY ternarymix
{Nfrac nfrac}
{isotherm | isobar |flashpoint}
End
nfracNumber of different mixtures for which the ternary mixture is calculated will be (nfrac+1)*(nfrac+2)/2. Default value for nfrac is 10, which means 55 different mixtures.
isotherm | isobar | flashpointIf the subkey isotherm is included (default) the ternary mixture will be calculated at a fixed temperature. If the subkey isobar is included the ternary mixture will be calculated at a fixed vapor pressure. If the subkey flashpoint is included the flash point of the ternary mixture will be calculated.
The input pure compound vapor pressure will be used in the calculation of the partial vapor pressure of this compound in the mixture if it is supplied with the key COMPOUND for this compound. If it is not specified then it will be approximated using the COSMO-RS method.
In case of a miscibility gap liquid-liquid equilibrium (LLE) data such as tie lines and an approximate phase diagram, are calculated. With the COSMO-RS GUI, activity coefficients, (partial) vapor pressures, and excess energies can be viewed as a colormap in a 2-dimensional plot with 2 of the liquid compositions on the axes.
Links COSMO-RS GUI tutorial: ternary mixtures (VLE/LLE) [1]
Solvents s1 - s2 Composition Line¶
The COSMO-RS module can linear interpolate between the compositions of solvent 1 and solvent 2, which both could be mixtures, and calculate properties, like activity coefficients, and excess energies. This property calculation does not calculate a possible miscibility gap. The mole fraction of each compound of the solvent 1 and solvent 2 should be given with the subkey FRAC1 and subkey FRAC2 of the key COMPOUND for each compound, respectively.
PROPERTY compositionline
{Nfrac nfrac}
{isotherm | isobar | flashpoint}
End
nfracNumber of different mixtures of the 2 solvents is calculated will be (nfrac+1). Default value for nfrac is 10, which means 11 different mixtures.
isotherm | isobar | flashpointIf the subkey isotherm is included (default) a fixed temperature will be used. If the subkey isobar is included a fixed vapor pressure will be used. If the subkey flashpoint is included the flashpoint will be calculated.
Links COSMO-RS GUI tutorial: A composition line between solvents s1 and s2 [1]
References