Modeling Electrodeposition and Dendrite Growth¶
Note
To follow this tutorial, either:
Download
Electrodeposition.py(run as$AMSBIN/amspython Electrodeposition.py).Download
Electrodeposition.ipynb(see also: how to install Jupyterlab)
Optionally:
Download
Electrodeposition-inlet.py.Download
Electrodeposition-inlet.ipynb.
In battery engineering, controlling how metal grows at the microscopic level is a critical safety requirement. If electrodeposition is uneven, it can form sharp, needle-like dendrites that grow across the electrolyte and cause massive internal short circuits. Therefore, understanding and suppressing this dendrite formation is essential to preventing catastrophic battery failure.
In this tutorial, we will build a minimal kinetic Monte Carlo (KMC) model for metal electrodeposition using pyZacros. By explicitly simulating individual atomic events (such as ion diffusion in the electrolyte, electrochemical reduction at the interface, and surface self-diffusion on the electrode), we can visualize how macroscopic morphologies develop from the electrode.
Inspired by the work of Vishnugopi et al. Surface diffusion manifestation in electrodeposition of metal anodes Phys. Chem. Chem. Phys., 2020,22, 11286-11295, the core goal of this tutorial is to show how the competition between different surface diffusion pathways dictates whether a deposited film grows smooth and flat, or rough and fractal. Here we describe a simplified model. For surface diffusion, we will specifically include terrace diffusion and step-detachment (diffusion away from a step edge). By the end, you will have run a short simulation and visualized the direct link between atomic-scale kinetics and the resulting electrodeposition morphology.
OK, let’s start!
First, we import the main packages we need:
import scm.pyzacros as pz
import math
Next, we configure our environment for reproducibility and
visualization. We set a deterministic random seed and import the
matplotlib tools needed to visualize the simulation results.
import random
random_seed = 100
random.seed(random_seed)
%matplotlib inline
import matplotlib.pyplot as plt
import matplotlib.animation as animation
plt.rcParams["figure.figsize"] = [7, 7]
from IPython.display import HTML
Now, define a convenient set of physical constants:
kB = 8.6173e-05 # Boltzmann constant in eV/K
F = 96487.0 # Faraday constant in C/mol
R = 8.314 # Gas constant in J/mol/K
N_a = 6.022e23 # Avogadro constant in 1/mol
To reproduce these morphological changes, this tutorial will guide you through the complete setup of the pyZacros simulation. We will start from the ground up by defining the species and their energetic cluster expansions. With the physical entities established, we will build a 2D lattice and populate it with our initial electrode surface and electrolyte ions. From there, we will encode the actual physics (ion transport, reduction, and surface diffusion) into a network of elemental reactions. Finally, we will wrap these components into a ZacrosJob calculation, execute the KMC simulation, and animate the results to watch our metal deposit grow dynamically over time.
Species and Energetics (Clusters)¶
We first need to define the fundamental building blocks of our system.
As shown in the diagram below, we allow each lattice site to take on one
of three states: an empty site * (representing the background
solvent), a solid deposited metal atom M, or a solvated metal cation
M+.

s = pz.Species("*", denticity=1, kind=pz.Species.SURFACE)
M = pz.Species("M", denticity=1, kind=pz.Species.SURFACE, mass=1.0)
Mp = pz.Species("M+", denticity=1, kind=pz.Species.SURFACE, mass=1.0)
Next, we use Clusters to define the energetics of these species. To keep
this model minimal, we will stick to one-site energetic terms for M
and M+ and assign them a baseline energy of zero. We also
intentionally omit lateral interactions. In pyZacros, this means our
cluster expansion is simply a single site (s) occupied by either an
M or M+ species.
M_cluster = pz.Cluster(label="M", site_types=["s"], species=[M])
Mp_cluster = pz.Cluster(label="Mp", site_types=["s"], species=[Mp])
The Lattice¶
Our first step is to construct a 2D square lattice with nearest-neighbor connectivity. We set the distance between adjacent sites to 3.5 Å (\(a = 3.5\)), which corresponds to the atomic spacing of our metal electrode. The lattice has periodic boundary conditions along both axes. Following the reference paper, we map the liquid electrolyte onto this same crystalline structure. We capture the physical difference between the liquid and solid phases indirectly by assigning different kinetic rates to their respective diffusion mechanisms. For the simulation size, we intentionally chose a 100 x 100 grid. This specific scale strikes the perfect balance: it lets us track individual atomic hops while providing a large enough canvas to clearly see macroscopic features like nucleation density, growth front roughness, and island coalescence.
The code below creates this lattice and plots it so we can verify the setup is correct before moving on.
a = 3.5 # Lattice constant in ang
lattice = pz.Lattice(
cell_vectors=[[a, 0.0], [0.0, a]],
repeat_cell=[100, 100],
site_types=["s"],
site_coordinates=[[0.000000, 0.000000]],
neighboring_structure=[[(0, 0), pz.Lattice.EAST], [(0, 0), pz.Lattice.NORTH]],
)
lattice.plot(marker_size=1, markers=["o"], colors=["0.8"]);

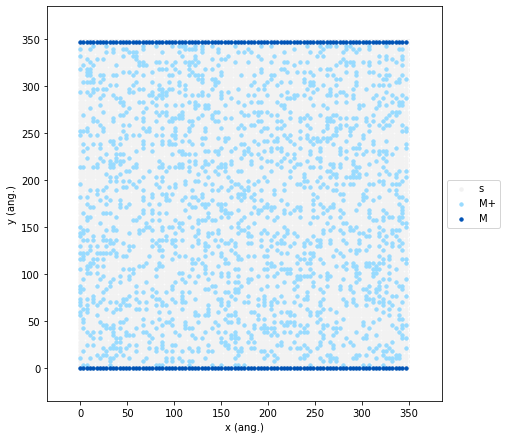
The Initial State¶
Once the lattice is built, we need to set up our initial “experimental”
conditions. First, we create a Growth Front. We seed a horizontal line
of M atoms along the bottom and top edges of the lattice. This acts
as our initial conductive electrode surface where deposition will begin.
Next, we add the electrolyte by randomly filling 20% of the remaining
lattice sites with M+ ions. This specific 20% concentration provides
a sufficient ionic flux to trigger visible competition between the two
key processes, the rate at which ions reduce onto the surface, and the
rate at which they diffuse along it.
initial_state = pz.LatticeState(lattice, [M, Mp, s], initial=True)
for sid in range(0, 100 * 100, 100):
initial_state.fill_site(sid, M)
initial_state.fill_site(sid - 1, M)
initial_state.fill_sites_random("s", Mp, 0.2)
0.2
To verify that our initial state is configured correctly, we can visualize it using the following code:
initial_state.plot(marker_size=2.5, markers=["o", "o"], colors=["#98dbff", "#0053b6"], lattice_color="0.95");
Elemental Reactions & Bulk Diffusion¶
The core of any KMC simulation is its reaction network. In our model, the electrodeposition dynamics are driven by four different elementary events:
Bulk Diffusion:
M+ions swap places with neighboring empty sites (*) to simulate random walk transport through the liquid electrolyte.Electrochemical Reduction: An
M+ion adjacent to a solidMatom receives an electron and is reduced into a newly depositedMatom.Surface Diffusion: Deposited
Matoms hop across the surface. We explicitly track two pathways: terrace diffusion (moving along a flat plane) and step-detachment (moving away from a step edge).Ion Replenishment: A background source term acts as an infinite reservoir, continuously introducing new
M+ions to maintain a steady concentration.
Now, let’s translate each of these physical steps into pyZacros elementary reactions.
Electrolyte Diffusion
First, let’s model how ions move through the liquid. We define ionic
electrolyte diffusion as a reversible two-site hop where an solvated
M+ ion swaps places with an adjacent empty site (*). On the left
side of the image below, there is a representation of this process using
graphs, as needed for pyZacros, which in this specific case consists of
only two nodes. On the right side is the enumeration of the graph’s
nodes. This graph is trivial, but it will be very useful later for more
complex reactions.

Because diffusion in a liquid is faster compared to solid-state surface
reactions, inputting raw physical rates introduces a computational
“stiffness” problem into our KMC model. The solver would get bogged down
simulating millions of fast ion hops without any actual deposition
happening. To fix this, we apply a diffusion scaling factor (sf).
This trick artificially slows down diffusion just enough to keep the
simulation efficient, while maintaining the physical rule that bulk
transport must remain at faster than the surface processes. For our
setup, a scaling factor of 0.1 preserves this physical hierarchy.
We derive our diffusion coefficient from Valoen and Reimers J. Electrochem. Soc. 152 A882. Their fitted data for LiPF\(_6\) electrolytes suggests that at high concentrations (>4M, as in our case) and around 300 K, a reasonable order-of-magnitude estimate is \(D = 5 \cdot 10^{-7}\) cm\(^2\)/s.
Finally, when translating this into the pyZacros elementary reaction,
notice that we set the activation energy to zero
(activation_energy = 0). Because Zacros calculates rates using the
Arrhenius equation, a zero activation barrier forces the exponential
term to 1. This ensures that the prefactor we supply becomes equal to
our rate constant.
D = 5.0e-7 # Diffusion coefficient of M+ in the electrolyte in cm^2/s
sf = 0.1 # Scaling diffusion factor
k_T = (D / a**2) * (1e16) # Ion diffusion rate in 1/s
MpDiffusion = pz.ElementaryReaction(
label="MpDiffusion",
site_types=["s", "s"],
initial=[Mp, s],
final=[s, Mp],
initial_entity_number=[0, 1],
final_entity_number=[0, 1],
neighboring=[(0, 1)],
reversible=True,
pre_expon=sf * k_T,
pe_ratio=1.0,
activation_energy=0.0,
)
Electrochemical Reduction
In this step, a local pair consisting of a solvated ion and a solid
metal atom ([Mp, M]) transforms into two solid metal atoms
([M, M]). This represents the critical electron transfer event at
the metal/electrolyte interface. Just as we did for bulk diffusion, the
image below breaks down this process. On the left side, we see the graph
representation of the physical reduction event. On the right side, we
have the corresponding node enumeration.

We start by calculating the overall reaction current density (\(J\))
using the classic Butler-Volmer equation. You will likely recognize the
standard experimental parameters used here, such as the applied
overpotential (\(\eta\), eta), the exchange current density
(\(i_0\)), and the operating temperature (\(T\)). However,
pyZacros does not work with macroscopic currents, it requires a specific
event frequency for a single atom! Therefore, we take \(J\) and
scale it down to the area of just one lattice site (\(a^2\)). After
applying standard unit conversions using Faraday’s constant (\(F\))
and Avogadro’s number (\(N_a\)), we finally arrive at \(k_R\),
the per-site reaction rate in events per second. Finally, just as we did
for bulk diffusion, we set activation_energy = 0, so our calculated
\(k_R\) plugs directly in as the pre_expon parameter.
eta = 0.6 # Local overpotential in eV
i_0 = 5.0 # Exchange current density in mA/cm^2
alpha = 0.7 # Charge transfer coefficient alpha
beta = 1.0 - alpha # Charge transfer coefficient beta
T = 300.0 # Operating temperature in K
J = i_0 * (
math.exp(alpha * F * eta / R / T) - math.exp(-beta * F * eta / R / T)
) # Faradaic reaction current density in mA/cm^2
k_R = J * a**2 * (N_a / F) * (1.0 / 1e3) * (1.0 / 1e16) # Electrochemical reaction rate in 1/s
MpReduction = pz.ElementaryReaction(
label="MpReduction",
site_types=["s", "s"],
initial=[Mp, M],
final=[M, M],
initial_entity_number=[0, 1],
final_entity_number=[0, 1],
neighboring=[(0, 1)],
reversible=False,
pre_expon=k_R,
activation_energy=0.0,
)
Surface Diffusion
Once an atom is reduced onto the electrode, it does not necessarily stay where it landed. It can hop to neighboring sites. We model this surface self-diffusion using standard Arrhenius hopping rates:
Here, \(\nu\) represents the attempt frequency—how often an atom vibrates and “tries” to jump to a new site (typically \(10^{12} - 10^{13}\) s\(^{-1}\)). For this example, we set \(\nu = 2 \cdot 10^{12}\) s\(^{-1}\) (nu)
nu = 2e12 # surface self-diffusion hopping frequency in 1/s
The activation barrier \(E_a\) will change depending on the local geometry of the hop. We use the values from the reference paper, but you can find inspiration on how to calculate such parameters from scratch using a quantum chemistry package (like the Amsterdam Modeling Suite) in this reference: Jäckle M. et al. Self-diffusion barriers: possible descriptors for dendrite growth in batteries? Energy Environ. Sci., 2018,11, 3400-3407.
To connect our model with the mechanistic discussion in the Vishnugopi et al. paper, we divide surface mobility into two distinct pathways.
Terrace diffusion: An atom hops laterally across a flat layer (intralayer hopping).
Diffusion away from a step: An atom escapes from a highly coordinated step-edge environment to a less coordinated site.
The original paper also studies a third pathway: interlayer (across-step) diffusion, where an atom descends from an upper terrace to a lower one. The authors identified this as the predominant mechanism for smoothing the surface and suppressing dendrites. To keep our tutorial model minimal and computationally fast, we have intentionally omitted this descending pathway.
First up is terrace diffusion. To implement this, we must translate the physical mechanism into a graph-based representation. The goal is to keep this representation as small as possible without losing the underlying physics (for instance, we need enough surrounding nodes to guarantee the atom is actually on a flat terrace and not perched on a step). The image below illustrates the diffusion process for our proposed graph representation and the corresponding node enumeration:

Based on the reference paper, we use \(E_a = 0.15\) eV for this reaction. The following code demonstrates how to translate the graph above into a pyZacros elementary reaction:
E_a1 = 0.15
MTerraceDiffusion = pz.ElementaryReaction(
label="MTerraceDiffusion",
# 0 , 1 , 2 , 3 , 4 , 5 , 6 , 7
site_types=["s", "s", "s", "s", "s", "s", "s", "s"],
initial=[s, s, s, M, s, s, M, M],
final=[s, s, s, s, M, s, M, M],
neighboring=[(0, 1), (0, 3), (1, 4), (2, 3), (3, 4), (3, 6), (4, 5), (4, 7), (6, 7)],
reversible=False,
pre_expon=nu,
activation_energy=E_a1,
)
Next, we define diffusion away from a step. Just as before, we translate this physical process into a graph. Notice that this specific mechanism requires a slightly larger 9-node cluster to properly capture the local geometry of the step and ensure the atom is moving away from it.

Following the reference paper, we assign this reaction an activation barrier of \(0.30\) eV. Notice that this is twice as high as the terrace diffusion barrier (\(0.15\) eV)! This energy penalty physically represents the difficulty of breaking the extra atomic bonds associated with a step-edge environment, capturing the reduced mobility of these trapped atoms.
Here is how we translate this graph into our final surface diffusion pyZacros reaction:
E_a2 = 0.30
MDiffusionAwayFromStep = pz.ElementaryReaction(
label="MDiffusionAwayFromStep",
# 0 , 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8
site_types=["s", "s", "s", "s", "s", "s", "s", "s", "s"],
initial=[s, s, M, M, s, s, M, M, M],
final=[s, s, M, s, M, s, M, M, M],
neighboring=[(0, 1), (0, 3), (1, 4), (2, 3), (2, 6), (3, 4), (3, 7), (4, 5), (4, 8), (6, 7), (7, 8)],
reversible=False,
pre_expon=nu,
activation_energy=E_a2,
)
Using a “Fictitious Gas” Strategy to Keep Ions Constant
To avoid the rapid depletion of M+ ions during deposition, we introduce
a numerical replenishment event. We use a fictitious gas species
(Mg) that continuously produces new M+ ions on empty sites at a
constant rate \(k_A\).

Assuming a scenario where diffusion in the electrolyte is much faster than the interfacial reduction step, the ion addition rate should depend directly on the surface reduction rate
Here, \(f_A\) can be physically interpreted as the probability
fraction of having an M+ ion sitting directly against the electrode,
ready to react. In practice, it serves as an empirical tuning parameter
adjusted by trial and error to keep the overall ion concentration
approximately constant. For our specific setup, we achieved this balance
by setting \(f_A = 0.0005\).
The following code adds this fictitious gas species and the replenishment mechanism to our simulation:
Mg = pz.Species("Mg", gas_energy=0.0, mass=1.0)
f_A = 0.0005
k_A = f_A * k_R
MpAddIons = pz.ElementaryReaction(
label="MpAddIons",
site_types=["s"],
initial=[s, Mg],
final=[Mp],
reversible=False,
pre_expon=k_A,
activation_energy=0.0,
)
Setting up the ZacrosJob and Running the Calculation¶
Now that we have all that we need, we are ready to configure the
calculation. We do this using a Settings object, which controls the
macroscopic conditions (temperature, pressure), simulation stopping
criteria, and sampling time for different properties. Notice that we set
the molar fraction of our fictitious gas (Mg) to 1.0. We also define
an output sampling interval (\(\Delta t = 3 \cdot 10^{-8}\) s) to
ensure we capture enough frames to properly resolve both the transient
growth dynamics and the final surface morphology.
sett = pz.Settings()
sett.temperature = T
sett.pressure = 1.0
sett.molar_fraction.Mg = 1.0
sett.random_seed = random_seed
dt = 3e-8
sett.max_time = 15 * dt
sett.snapshots = ("time", dt)
sett.process_statistics = ("time", dt)
sett.species_numbers = ("time", 0.25 * dt)
Finally, we pack all components into a single ZacrosJob and execute it. Grab a coffee! On a typical laptop, this simulation should take about 20 minutes to complete!
cluster_expansion = [M_cluster, Mp_cluster]
mechanism = [MpDiffusion, MpReduction, MTerraceDiffusion, MDiffusionAwayFromStep, MpAddIons]
job = pz.ZacrosJob(
settings=sett,
lattice=lattice,
mechanism=mechanism,
cluster_expansion=cluster_expansion,
initial_state=initial_state,
)
results = job.run()
[31.03|10:47:07] JOB plamsjob STARTED
[31.03|10:47:09] JOB plamsjob RUNNING
[31.03|11:06:04] JOB plamsjob FINISHED
[31.03|11:06:04] JOB plamsjob SUCCESSFUL
Analyzing the Results¶
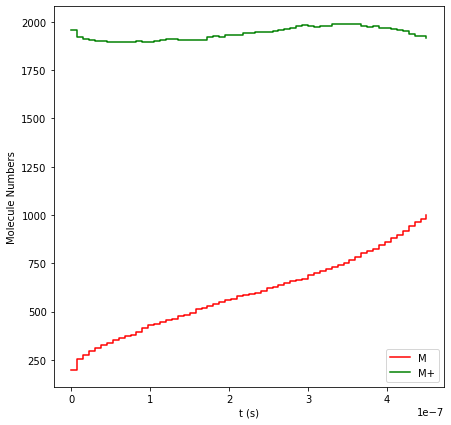
First, let’s look at how the species populations evolve over time. By
plotting the molecule numbers, we can verify that our fictitious gas
trick worked! The concentration of solvated M+ ions (the green line)
remains approximately constant throughout the entire simulation. Notice
the gradual deposition of solid M atoms (the red line), steadily
growing from the initial seed surface up to a total of about 1000 atoms.
results.plot_molecule_numbers(["M", "M+"]);
To inspect the final spatial distribution of our deposited film, we can
plot the last frame of the lattice_states trajectory (using index
[-1]):
results.plot_lattice_states(
results.lattice_states()[-1],
marker_size=2.5,
markers=["o", "o"],
colors=["#98dbff", "#0053b6"],
lattice_color="0.95",
);
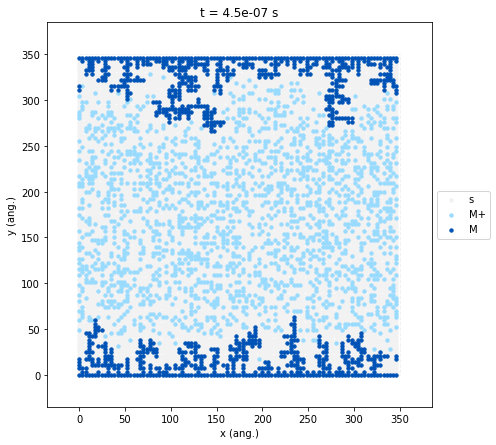
The resulting plot reveals the macroscopic morphology of the
electrodeposited metal (M, dark blue). As expected from our kinetic
parameters, the growth fronts have advanced inward from the boundaries,
but the resulting deposit is highly uneven, exhibiting severe roughening
and dendritic branching. This morphology is a direct consequence of our
chosen reaction network. The fast electrochemical reduction rate
outpaces the lateral surface diffusion (terrace diffusion and
step-detachment). Furthermore, because we intentionally excluded
interlayer (descending) diffusion from this minimal model, deposited
atoms cannot easily relax into lower layers. This forces the system to
grow outward into peaks and branches rather than forming a dense,
conformal film.
As a final step, we can visualize the entire deposition process as a
continuous animation. This dynamic view is often much more informative
than a single final snapshot when trying to diagnose which kinetic step
limits morphology evolution. The following cell uses
matplotlib.animation.ArtistAnimation to compile our sequence of
lattice states into a movie. We then use the to_jshtml() method to
render an interactive HTML5 video player.
fig, ax = plt.subplots()
frames = []
results.plot_lattice_states(
results.lattice_states(),
show=False,
marker_size=2.5,
markers=["o", "o"],
colors=["#98dbff", "#0053b6"],
lattice_color="0.95",
frames=frames,
)
ani = animation.ArtistAnimation(fig, frames, interval=500, blit=True)
plt.close(fig) # prevent extra static figure display in notebook
HTML(ani.to_jshtml())
Controlling Morphology: The Low Overpotential Regime¶
You can easily alter the final morphology by shifting the kinetic
balance of the simulation. If you adjust the initial parameters to a
lower overpotential (eta = 0.4) and a longer time step
(dt = 1e-6) to account for the slower reactions, the simulation will
produce a dense, conformal film rather than dendritic branches.

Achieving this film-type deposition is the ultimate goal in metal anode engineering. A flat, uniform morphology minimizes localized current density “hot spots”, extends battery cycle life, and eliminates the safety hazards associated with dendrite-induced short circuits.
This shift in growth mode happens because lowering the overpotential exponentially decreases the rate of electrochemical reduction. With a much slower influx of new solid atoms, the system is no longer diffusion-limited. The deposited atoms have ample time to undergo lateral surface diffusion, allowing them to reorganize and relax into highly coordinated, tightly packed configurations before subsequent layers are formed.
The Inlet Model: Simulating Diffusion-Limited Growth (Optional)¶
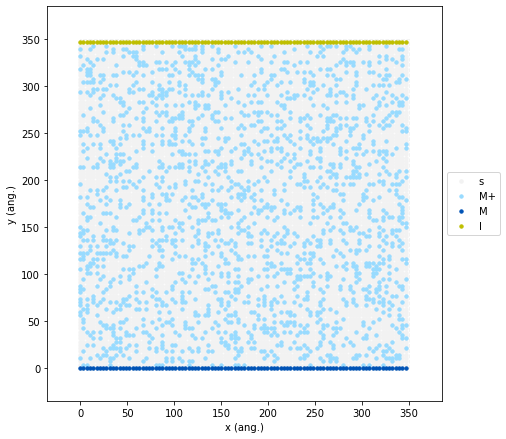
We can also model a system with a distinct concentration gradient by defining an explicit ion inlet rather than a uniform background reservoir.
In the code below, we define a new species I (Inlet). We initialize the lattice with our standard solid M electrode at the bottom (dark blue) and a solid line of I sites at the top (yellow). We also keep our initial 20% random seeding of M+ ions in the electrolyte solution.
I = pz.Species("I", denticity=1, kind=pz.Species.SURFACE, mass=1.0)
I_cluster = pz.Cluster(label="I", site_types=["s"], species=[I])
initial_state = pz.LatticeState(lattice, [M, Mp, s], initial=True)
for sid in range(0, 100 * 100, 100):
initial_state.fill_site(sid, M)
initial_state.fill_site(sid - 1, I)
initial_state.fill_sites_random("s", Mp, 0.2)
initial_state.plot(marker_size=2.5, markers=["o", "o", "o"], colors=["#98dbff", "#0053b6", "y"], lattice_color="0.95");
In this geometry, new M+ ions are continuously pumped into the
system by the top yellow layer. This means they must physically diffuse
across the entire simulation box to reach the bottom electrode before
they can be reduced.
To implement this, we define a new elementary reaction where our
fictitious Mg gas can only generate an M+ ion on an empty site
(s) if it is directly adjacent to an Inlet site (I). Notice that
we also increased our scaling fraction to f_A = 0.02. Because ions
are now only entering the system from a single boundary row rather than
uniformly across the whole grid, we need a higher local injection rate
to maintain a steady overall flux.
Mg = pz.Species("Mg", gas_energy=0.0, mass=1.0)
f_A = 0.02
k_A = f_A * k_R
MpAddIons = pz.ElementaryReaction(
label="MpAddIons",
site_types=["s", "s"],
initial=[I, s, Mg],
final=[I, Mp],
neighboring=[(0, 1)],
reversible=False,
pre_expon=k_A,
activation_energy=0.0,
)
Finally, we update our cluster_expansion to include the new inlet
species, our mechanism with the new MpAddIons reaction and
execute the updated ZacrosJob. To ensure the injected M+ ions have
enough time to diffuse from the top boundary down to the working
electrode, we increased the time step by a factor of 4 (previously
\(3\cdot 10^{-8}\) s), which indirectly increased the total
simulation time. Perfect time for a lunch break! On a typical laptop,
this calculation will take around an hour to wrap up.
cluster_expansion = [M_cluster, Mp_cluster, I_cluster]
mechanism = [MpDiffusion, MpReduction, MTerraceDiffusion, MDiffusionAwayFromStep, MpAddIons]
dt = 1.2e-7
sett.max_time = 15 * dt
sett.snapshots = ("time", dt)
sett.process_statistics = ("time", dt)
sett.species_numbers = ("time", 0.25 * dt)
job = pz.ZacrosJob(
settings=sett,
lattice=lattice,
mechanism=mechanism,
cluster_expansion=cluster_expansion,
initial_state=initial_state,
)
results = job.run()
[31.03|11:10:31] JOB plamsjob STARTED
[31.03|11:10:33] JOB plamsjob RUNNING
[31.03|12:13:49] JOB plamsjob FINISHED
[31.03|12:13:49] JOB plamsjob SUCCESSFUL
We have successfully run our directional model! First, let’s take a look to the population numbers:
results.plot_molecule_numbers(["M", "M+"]);
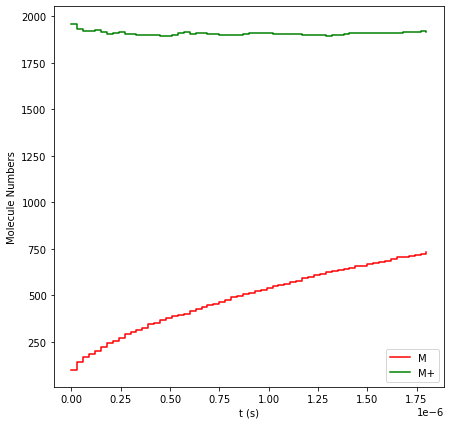
The green line shows that our new injection rate (f_A = 0.02)
perfectly balances the reduction rate, keeping the overall M+
concentration steady while the solid M film grows.
Now, lets see the final spatial distribution of our deposited film:
results.plot_lattice_states(
results.lattice_states()[-1],
marker_size=2.5,
markers=["o", "o", "o"],
colors=["#98dbff", "#0053b6", "y"],
lattice_color="0.95",
);
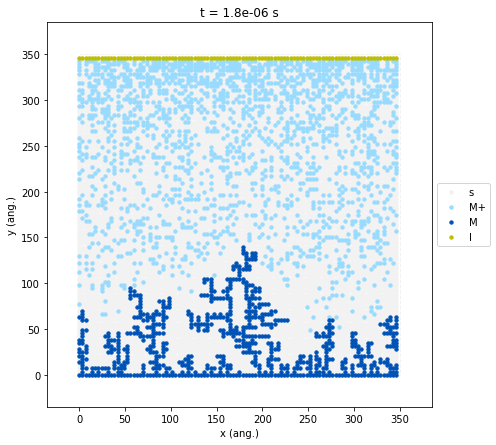
By introducing a physical distance between the ion source (the I
inlet at the top) and the reduction surface (the electrode at the
bottom), the simulation naturally develops a concentration gradient. The
M+ species (light blue) is highly concentrated near the inlet but
significantly depleted near the growth front. Furthermore, we can
clearly observe localized depletion zones around the dendrite tips. This
indicates a diffusion-limited growth regime. The highest peaks grow so
fast that they consume all the nearby ions before those ions can travel
down into the lower areas. Because the valleys are left “starved”, an
even film cannot form, which just causes the dendrites to grow further
out of control.
Now, just for fun, let’s create an animation to watch the dendritic morphology evolve over time!
fig, ax = plt.subplots()
frames = []
results.plot_lattice_states(
results.lattice_states(),
show=False,
marker_size=2.5,
markers=["o", "o", "o"],
colors=["#98dbff", "#0053b6", "y"],
lattice_color="0.95",
frames=frames,
)
ani = animation.ArtistAnimation(fig, frames, interval=500, blit=True)
plt.close(fig) # prevent extra static figure display in notebook
HTML(ani.to_jshtml())