Water-Gas Shift Reaction on Pt(111)¶
This tutorial shows how to translate a typical Zacros workflow to pyZacros. To do this, we will look at the water-gas shift reaction on Pt(111). The required physical/chemical descriptions of the system have been published in the literature.
This example shows how to include gas species, transition states, as well as stable surface species, and the lateral interactions between them. All information about the energetics was obtained via density functional theory (DFT) calculations. Importantly, lateral interactions are also discussed and included in the example script.
The script can be downloaded through this link: WaterGasShiftOnPt111.py
The following section will discuss the structure of the script step-by-step. You can also skip ahead to check out the results of the simulation.
pyZacros Setup¶
A pyZacros script always starts by loading the pyZacros module. For this example, we also define the pz alias in order to quickly access the necessary functions.
1import scm
2import scm.pyzacros as pz
We then proceed by defining the chemical species that make up the reaction system. This is done using the Species API. By default, the reference energy is set to 0. By including an * in the name of a species, they are automatically designated as an adsorbate.
1# ---------------------------------------------
2# Species:
3# ---------------------------------------------
4# - Gas-species:
5CO_gas = pz.Species("CO")
6H2O_gas = pz.Species("H2O")
7H2_gas = pz.Species("H2")
8CO2_gas = pz.Species("CO2", gas_energy=-0.615)
9O2_gas = pz.Species("O2", gas_energy=4.913)
10
11# - Surface species:
12s0 = pz.Species("*", 1) # Empty adsorption site
13CO_adsorbed = pz.Species("CO*", 1)
14H2O_adsorbed = pz.Species("H2O*", 1)
15OH_adsorbed = pz.Species("OH*", 1)
16O_adsorbed = pz.Species("O*", 1)
17H_adsorbed = pz.Species("H*", 1)
18COOH_adsorbed = pz.Species("COOH*", 1)
For constructing the lattice, we will use the built-in hexagonal lattice provided by pyZacros:
1# ---------------------------------------------
2# Lattice setup:
3# ---------------------------------------------
4latt = pz.Lattice(lattice_type=pz.Lattice.HEXAGONAL, lattice_constant=1.0, repeat_cell=[8, 10])
Now that we have a lattice, we can proceed by writing down the adsorption energies and the lateral interactions. For this example, we will limit ourselves to a small selection of nearest-neighbor interactions. Our set of cluster energies is then collected in a ClusterExpansion. This is a special Python list that allows us to more conveniently access the cluster energies.
1# ---------------------------------------------
2# Clusters:
3# ---------------------------------------------
4CO_point = pz.Cluster(species=[CO_adsorbed], energy=-2.077, label="CO_point")
5H2O_point = pz.Cluster(species=[H2O_adsorbed], energy=-0.362, label="H2O_point")
6OH_point = pz.Cluster(species=[OH_adsorbed], energy=0.830, label="OH_point")
7O_point = pz.Cluster(species=[O_adsorbed], energy=1.298, label="O_point")
8H_point = pz.Cluster(species=[H_adsorbed], energy=-0.619, label="H_point")
9COOH_point = pz.Cluster(species=[COOH_adsorbed], energy=-1.487, label="COOH_point")
10
11CO_pair_1NN = pz.Cluster(species=[CO_adsorbed, CO_adsorbed], neighboring=[(0, 1)], energy=0.560, label="CO_pair_1NN")
12OH_H_1NN = pz.Cluster(species=[OH_adsorbed, H_adsorbed], neighboring=[(0, 1)], energy=0.021, label="OH_H_1NN")
13O_H_1NN = pz.Cluster(species=[O_adsorbed, H_adsorbed], neighboring=[(0, 1)], energy=0.198, label="O_H_1NN")
14CO_OH_1NN = pz.Cluster(species=[CO_adsorbed, OH_adsorbed], neighboring=[(0, 1)], energy=0.066, label="CO_OH_1NN")
15CO_O_1NN = pz.Cluster(species=[CO_adsorbed, O_adsorbed], neighboring=[(0, 1)], energy=0.423, label="CO_O_1NN")
16
17# ---------------------------------------------
18# Cluster expansion:
19# ---------------------------------------------
20myClusterExpansion = pz.ClusterExpansion()
21myClusterExpansion.extend([CO_point, H2O_point])
22myClusterExpansion.append(OH_point)
23myClusterExpansion.extend([O_point, H_point, COOH_point])
24myClusterExpansion.extend([CO_pair_1NN, OH_H_1NN, O_H_1NN, CO_OH_1NN, CO_O_1NN])
The reaction mechanism is constructed by writing out the elementary steps. For each reaction, we define the reactants and products, the reversibility, and the kinetic parameters. (Note how the parameter names match those found in the Zacros input files.)
1# ---------------------------------------------
2# Elementary Reactions
3# ---------------------------------------------
4CO_adsorption = pz.ElementaryReaction(
5 initial=[s0, CO_gas],
6 final=[CO_adsorbed],
7 reversible=True,
8 pre_expon=2.226e007,
9 pe_ratio=2.137e-006,
10 activation_energy=0.0,
11 label="CO_adsorption",
12)
13
14H2_dissoc_adsorp = pz.ElementaryReaction(
15 initial=[s0, s0, H2_gas],
16 final=[H_adsorbed, H_adsorbed],
17 neighboring=[(0, 1)],
18 reversible=True,
19 pre_expon=8.299e007,
20 pe_ratio=7.966e-006,
21 activation_energy=0.0,
22 label="H2_dissoc_adsorp",
23)
24
25H2O_adsorption = pz.ElementaryReaction(
26 initial=[s0, H2O_gas],
27 final=[H2O_adsorbed],
28 reversible=True,
29 pre_expon=2.776e002,
30 pe_ratio=2.665e-006,
31 activation_energy=0.0,
32 label="H2O_adsorption",
33)
34
35H2O_dissoc_adsorp = pz.ElementaryReaction(
36 initial=[H2O_adsorbed, s0],
37 final=[OH_adsorbed, H_adsorbed],
38 neighboring=[(0, 1)],
39 reversible=True,
40 pre_expon=1.042e13,
41 pe_ratio=1.000e00,
42 activation_energy=0.777,
43 label="H2O_dissoc_adsorp",
44)
45
46OH_decomposition = pz.ElementaryReaction(
47 initial=[s0, OH_adsorbed],
48 final=[O_adsorbed, H_adsorbed],
49 neighboring=[(0, 1)],
50 reversible=True,
51 pre_expon=1.042e13,
52 pe_ratio=1.000e00,
53 activation_energy=0.940,
54 label="OH_decomposition",
55)
56
57COOH_formation = pz.ElementaryReaction(
58 initial=[CO_adsorbed, OH_adsorbed],
59 final=[s0, COOH_adsorbed],
60 neighboring=[(0, 1)],
61 reversible=True,
62 pre_expon=1.042e13,
63 pe_ratio=1.000e00,
64 activation_energy=0.405,
65 label="COOH_formation",
66)
67
68COOH_decomposition = pz.ElementaryReaction(
69 initial=[COOH_adsorbed, s0],
70 final=[s0, H_adsorbed, CO2_gas],
71 neighboring=[(0, 1)],
72 reversible=False,
73 pre_expon=1.042e13,
74 activation_energy=0.852,
75 label="COOH_decomposition",
76)
77
78CO_oxidation = pz.ElementaryReaction(
79 initial=[CO_adsorbed, O_adsorbed],
80 final=[s0, s0, CO2_gas],
81 neighboring=[(0, 1)],
82 reversible=False,
83 pre_expon=1.042e13,
84 activation_energy=0.988,
85 label="CO_oxidation",
86)
87
88# ---------------------------------------------
89# Full mechanism:
90# ---------------------------------------------
91mech = pz.Mechanism(
92 [
93 CO_adsorption,
94 H2_dissoc_adsorp,
95 H2O_adsorption,
96 H2O_dissoc_adsorp,
97 OH_decomposition,
98 COOH_formation,
99 COOH_decomposition,
100 CO_oxidation
101 ]
102)
Lastly, we define the general simulation settings. By creating a Settings object, we automatically load the default Zacros parameters. For this tutorial, we will use a short simulation time in order to quickly see the results of our simulation.
1# ---------------------------------------------
2# Settings:
3# ---------------------------------------------
4sett = pz.Settings()
5
6sett.molar_fraction.CO = 1.0e-5
7sett.molar_fraction.H2O = 0.950
8
9sett.random_seed = 123278
10sett.temperature = 500.0
11sett.pressure = 10.0
12sett.snapshots = ("time", 5.0e-4)
13sett.process_statistics = ("time", 5.0e-4)
14sett.species_numbers = ("time", 5.0e-4)
15sett.event_report = "off"
16sett.max_steps = "infinity"
17sett.max_time = 0.25
When we want to run a pyZacros job, we first start up Zacros itself by using the init() call. We then collect all of our settings into a ZacrosJob. The example script uses the print function to now show you an overview of the final settings, using the familiar Zacros input format (shown below).
The run function is used to start the actual Zacros simulation. Once the job has finished, we can use one of the built-in post-processing functions to plot the transient profiles for our key reaction species.
1scm.pyzacros.init()
2
3job = pz.ZacrosJob(settings=sett, lattice=latt, mechanism=mech, cluster_expansion=myClusterExpansion)
4
5print(job)
6results = job.run()
7
8if job.ok():
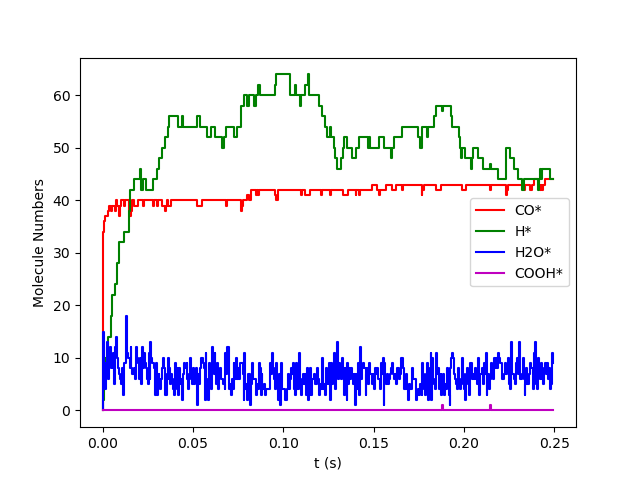
9 results.plot_molecule_numbers(["CO*", "H*", "H2O*", "COOH*"])
10
11scm.pyzacros.finish()
The finish() call is used to wrap up the Zacros job and close the script.
Running the Simulation¶
pyZacros scripts are executed using PLAMS. The PLAMS documentation contains examples for running Python workflows on Windows, Linux and MacOS systems. If you are using the default AMS installation:
$AMSBIN/amspython WaterGasShiftOnPt111.py
The script will first print an overview of the provided simulation parameters.
The actual simulation (denoted by the plamsjob) will then start running. This example script only runs for a very short time, and should finish in under 1 minute.
A figure will be shown at the end of the simulation, containing the transient composition of surface species.
1$ amspython WaterGasShiftOnPt111.py
2PLAMS working folder: /home/user/pyzacros/examples/WaterGasShiftOnPt111/plams_workdir
3---------------------------------------------------------------------
4simulation_input.dat
5---------------------------------------------------------------------
6random_seed 123278
7temperature 500.0
8pressure 10.0
9
10snapshots on time 0.0005
11process_statistics on time 0.0005
12species_numbers on time 0.0005
13event_report off
14max_steps infinity
15max_time 0.25
16
17n_gas_species 4
18gas_specs_names CO H2 H2O CO2
19gas_energies 0.00000e+00 0.00000e+00 0.00000e+00 -6.15000e-01
20gas_molec_weights 2.79949e+01 2.01560e+00 1.80105e+01 4.39898e+01
21gas_molar_fracs 1.00000e-05 0.00000e+00 9.50000e-01 0.00000e+00
22
23n_surf_species 6
24surf_specs_names CO* H* H2O* OH* O* COOH*
25surf_specs_dent 1 1 1 1 1 1
26
27finish
28---------------------------------------------------------------------
29lattice_input.dat
30---------------------------------------------------------------------
31lattice default_choice
32 hexagonal_periodic 1.0 8 10
33end_lattice
34---------------------------------------------------------------------
35energetics_input.dat
36---------------------------------------------------------------------
37energetics
38
39cluster CO_point
40 sites 1
41 lattice_state
42 1 CO* 1
43 site_types 1
44 graph_multiplicity 1
45 cluster_eng -2.07700e+00
46end_cluster
47
48cluster H2O_point
49 sites 1
50 lattice_state
51 1 H2O* 1
52 site_types 1
53 graph_multiplicity 1
54 cluster_eng -3.62000e-01
55end_cluster
56
57cluster OH_point
58 sites 1
59 lattice_state
60 1 OH* 1
61 site_types 1
62 graph_multiplicity 1
63 cluster_eng 8.30000e-01
64end_cluster
65
66cluster O_point
67 sites 1
68 lattice_state
69 1 O* 1
70 site_types 1
71 graph_multiplicity 1
72 cluster_eng 1.29800e+00
73end_cluster
74
75cluster H_point
76 sites 1
77 lattice_state
78 1 H* 1
79 site_types 1
80 graph_multiplicity 1
81 cluster_eng -6.19000e-01
82end_cluster
83
84cluster COOH_point
85 sites 1
86 lattice_state
87 1 COOH* 1
88 site_types 1
89 graph_multiplicity 1
90 cluster_eng -1.48700e+00
91end_cluster
92
93cluster CO_pair_1NN
94 sites 2
95 neighboring 1-2
96 lattice_state
97 1 CO* 1
98 2 CO* 1
99 site_types 1 1
100 graph_multiplicity 1
101 cluster_eng 5.60000e-01
102end_cluster
103
104cluster OH_H_1NN
105 sites 2
106 neighboring 1-2
107 lattice_state
108 1 OH* 1
109 2 H* 1
110 site_types 1 1
111 graph_multiplicity 1
112 cluster_eng 2.10000e-02
113end_cluster
114
115cluster O_H_1NN
116 sites 2
117 neighboring 1-2
118 lattice_state
119 1 O* 1
120 2 H* 1
121 site_types 1 1
122 graph_multiplicity 1
123 cluster_eng 1.98000e-01
124end_cluster
125
126cluster CO_OH_1NN
127 sites 2
128 neighboring 1-2
129 lattice_state
130 1 CO* 1
131 2 OH* 1
132 site_types 1 1
133 graph_multiplicity 1
134 cluster_eng 6.60000e-02
135end_cluster
136
137cluster CO_O_1NN
138 sites 2
139 neighboring 1-2
140 lattice_state
141 1 CO* 1
142 2 O* 1
143 site_types 1 1
144 graph_multiplicity 1
145 cluster_eng 4.23000e-01
146end_cluster
147
148end_energetics
149---------------------------------------------------------------------
150mechanism_input.dat
151---------------------------------------------------------------------
152mechanism
153
154reversible_step CO_adsorption
155 gas_reacs_prods CO -1
156 sites 1
157 initial
158 1 * 1
159 final
160 1 CO* 1
161 site_types 1
162 pre_expon 2.22600e+07
163 pe_ratio 2.13700e-06
164 activ_eng 0.00000e+00
165end_reversible_step
166
167reversible_step H2_dissoc_adsorp
168 gas_reacs_prods H2 -1
169 sites 2
170 neighboring 1-2
171 initial
172 1 * 1
173 2 * 1
174 final
175 1 H* 1
176 2 H* 1
177 site_types 1 1
178 pre_expon 8.29900e+07
179 pe_ratio 7.96600e-06
180 activ_eng 0.00000e+00
181end_reversible_step
182
183reversible_step H2O_adsorption
184 gas_reacs_prods H2O -1
185 sites 1
186 initial
187 1 * 1
188 final
189 1 H2O* 1
190 site_types 1
191 pre_expon 2.77600e+02
192 pe_ratio 2.66500e-06
193 activ_eng 0.00000e+00
194end_reversible_step
195
196reversible_step H2O_dissoc_adsorp
197 sites 2
198 neighboring 1-2
199 initial
200 1 H2O* 1
201 2 * 1
202 final
203 1 OH* 1
204 2 H* 1
205 site_types 1 1
206 pre_expon 1.04200e+13
207 pe_ratio 1.00000e+00
208 activ_eng 7.77000e-01
209end_reversible_step
210
211reversible_step OH_decomposition
212 sites 2
213 neighboring 1-2
214 initial
215 1 * 1
216 2 OH* 1
217 final
218 1 O* 1
219 2 H* 1
220 site_types 1 1
221 pre_expon 1.04200e+13
222 pe_ratio 1.00000e+00
223 activ_eng 9.40000e-01
224end_reversible_step
225
226reversible_step COOH_formation
227 sites 2
228 neighboring 1-2
229 initial
230 1 CO* 1
231 2 OH* 1
232 final
233 1 * 1
234 2 COOH* 1
235 site_types 1 1
236 pre_expon 1.04200e+13
237 pe_ratio 1.00000e+00
238 activ_eng 4.05000e-01
239end_reversible_step
240
241step COOH_decomposition
242 gas_reacs_prods CO2 1
243 sites 2
244 neighboring 1-2
245 initial
246 1 COOH* 1
247 2 * 1
248 final
249 1 * 1
250 2 H* 1
251 site_types 1 1
252 pre_expon 1.04200e+13
253 activ_eng 8.52000e-01
254end_step
255
256step CO_oxidation
257 gas_reacs_prods CO2 1
258 sites 2
259 neighboring 1-2
260 initial
261 1 CO* 1
262 2 O* 1
263 final
264 1 * 1
265 2 * 1
266 site_types 1 1
267 pre_expon 1.04200e+13
268 activ_eng 9.88000e-01
269end_step
270
271end_mechanism
272[08.02|15:34:32] JOB plamsjob STARTED
273[08.02|15:34:32] JOB plamsjob RUNNING
274[08.02|15:34:57] JOB plamsjob FINISHED
275[08.02|15:34:57] JOB plamsjob SUCCESSFUL
276[08.02|15:35:10] PLAMS run finished. Goodbye