Quantum ESPRESSO: Calculating IR and Raman Spectra¶
Infrared (IR) and Raman spectroscopy are powerful techniques used to get information about the vibrational modes of molecules and solids and also provide valuable insights into the structure, bonding, and dynamics of materials. IR spectroscopy measures the absorption of infrared light, which excites vibrational modes that have a change in dipole moment. On the other hand, Raman spectroscopy measures the scattering of light, which excites vibrational modes that have a change in polarizability. By analyzing the frequencies and intensities of the absorbed or scattered light, we can identify the vibrational modes present in the material and gain information about its properties.
Beryllium oxide (BeO), also known as bromellite, is a wide band gap (7.46 eV) semiconductor. It has a wurtzite crystal structure (space group \(P6_3mc\), No. 186) and exhibits interesting optical properties, making it relevant for applications in catalysis, high-power electronics, and neutron moderators in nuclear reactors. We obtained the system’s geometry from The Materials Project (ID: mp-2542). From the computational point of view, this system is relatively simple (only 4 atoms per unit cell) and has a small number of electrons (20 electrons per unit cell), which leads to faster calculations, making it suitable for illustrative purposes. BeO has a hexagonal structure (\(C_{6v}\) (6mm) symmetry). Some vibrational modes are active in both IR and Raman spectroscopy, while others are only Raman active. This selectivity arises from the relationship between the symmetry of the vibrational mode and the selection rules governing IR and Raman activity. Theoretical results for comparison can be found in The Computational Raman Database (mpid: mp-2542), and the experimental Raman spectrum is available in the RRUFF Project (RRUFF ID: X050194).
Tip
The following paper is a highly recommended resource for anyone performing first-principles Raman spectroscopy calculations. It provides a solid foundation and practical guidance for accurate and efficient computations, complemented by a valuable database of theoretical results and corresponding experimental data.
G. Petretto, S. Dwaraknath, H. P.C. Miranda, et al. High-throughput density-functional perturbation theory phonons for inorganic materials. Sci Data 5, 180065 (2018). https://doi.org/10.1038/s41597-023-01988-5
Computational Methodology¶
The calculation of IR and Raman spectra using Quantum Espresso (QE) involves a series of steps that are handled internally by AMS, making the process transparent to the user. These steps use the QE tools pw.x, ph.x, and dynmat.x. For detailed information about these tools, refer to the official Quantum Espresso documentation online.
Starting from an optimized geometry, a self-consistent field (SCF) calculation is performed using pw.x to determine the electronic ground state of the system. This step solves the Kohn-Sham equations iteratively to obtain the converged electron density and ground state energy. Next, ph.x is employed to calculate the phonon frequencies and eigenvectors at a specific q-point (typically \(\Gamma\) point) in the Brillouin zone using density functional perturbation theory (DFPT) to compute the dynamical matrix, which captures the interatomic force constants. Finally, dynmat.x is used to post-process the phonon data, obtaining the IR and Raman intensities, frequencies, and vibrational modes. This step involves computing the Born effective charges and the Raman tensors, which describe the coupling of the vibrational modes to the electric field and the change in polarizability, respectively. These quantities are essential for determining the IR and Raman activity of the vibrational modes.
IR and Raman spectra can be calculated using Quantum Espresso through AMS via two main interfaces: the graphical user interface (GUI) and the Python library, PLAMS. The GUI presents a user-friendly, visual approach that is ideal for users performing routine calculations on simple systems. It simplifies the process of setting up input files and visualizing results. On the other hand, PLAMS offers a powerful and flexible Python environment suitable for complex workflows, parameter sweeps, and integration with other scientific libraries. This makes it particularly advantageous for advanced users, high-throughput calculations, or situations that require automation and scripting, providing greater control and customization of the computational process. In this tutorial, we will explore both options. They are completely independent, so you can jump directly to the section that interests you the most.
Using the GUI¶
We will use the following structure as basis for the subsequent calculations:
Be 0.0000000000 1.5555413800 0.0007009000
Be 1.3471383600 0.7777706900 2.1887103500
O 0.0000000000 1.5555413800 1.6512462200
O 1.3471383600 0.7777706900 3.8392556700
VEC1 2.6942767100 0.0000000000 0.0000000000
VEC2 -1.3471383600 2.3333120800 0.0000000000
VEC3 0.0000000000 0.0000000000 4.3760188900
Hint
To build a custom crystal, check out the Tutorial on Building Crystals and Slabs
Setting Up the Calculation:

You should get something like this:
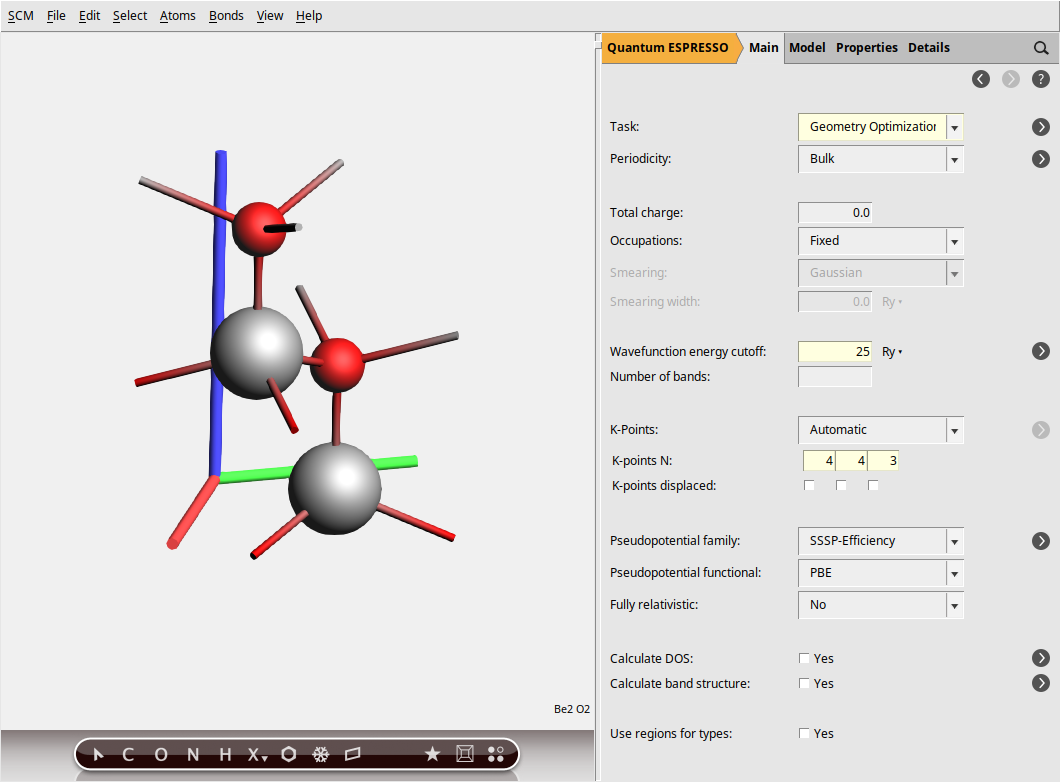
In the steps above, we specified a GeometryOptimization task to ensure our BeO structure is at a local energy minimum, preventing any spurious imaginary frequencies in the phonon spectrum. Next, for the electronic structure part, we set the plane-wave energy cutoff (ecutwfc) to 25 Ry. While this value is lower than typically recommended for production calculations (40 Ry), we use it here for demonstration purposes to reduce computational cost. Then, the Brillouin zone sampling is done using a Monkhorst-Pack k-point grid of size 4 × 4 × 3, ensuring a balanced sampling in all directions.
To specify the pseudopotentials, go to the Pseudopotentials configuration panel by selecting Model → Pseudopotentials. In this panel, use the file selection areas next to each element under “Pseudopotentials used:”. Clicking on the folder icon will open a file browser window. Navigate to the “QE” directory and select the appropriate pseudopotential file for each element.
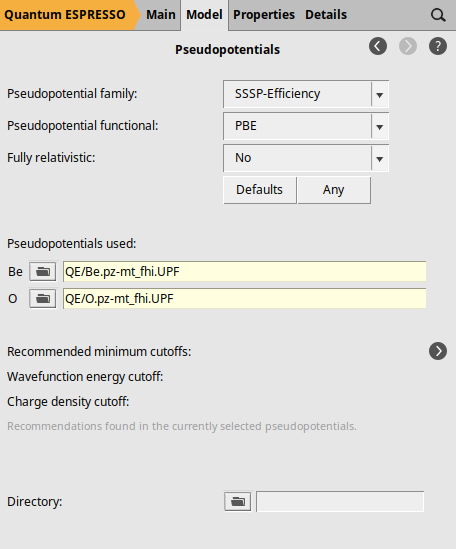
Notice that we have specified the pseudopotentials by selecting the files directly instead of using a pseudopotential family. This is due to Quantum Espresso’s limitation of only supporting Norm-Conserving (NC) and LDA-type pseudopotentials for Raman spectra calculations, which are not included in our standard pseudopotential families. For this example, we selected the pseudopotential files: Be.pz-mt_fhi.UPF for beryllium and O.pz-mt_fhi.UPF for oxygen. The filenames of these pseudopotentials indicate that they use the Perdew-Zunger LDA functional (pz), the Martins-Troullier generation method (mt), and that they originate from the FHI-aims library (fhi).
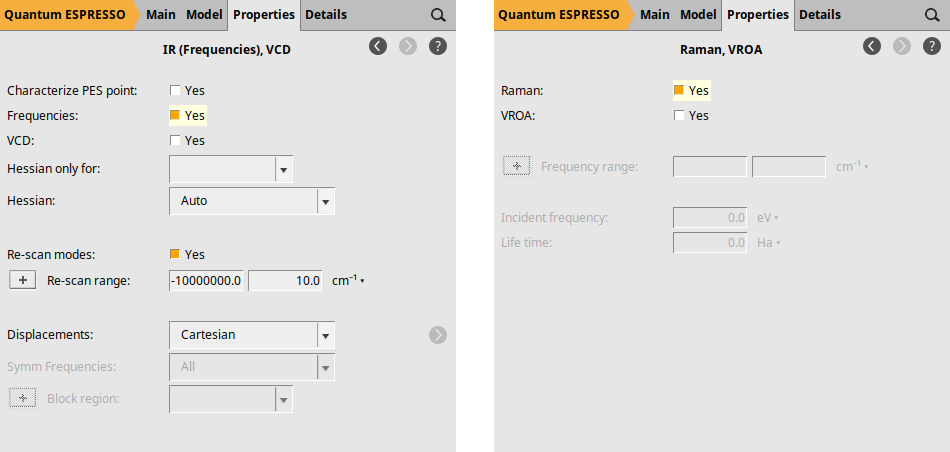
Finally, we explicitly request the calculation of NormalModes (which yields the IR spectrum) and the Raman spectrum. To request the IR spectrum, go to Properties → IR (Frequencies), VCD and check the box next to Frequencies: by selecting Yes. For the Raman spectrum, navigate to Properties → Raman, VROA and select Yes in the Raman: checkbox.
Computation & Results¶
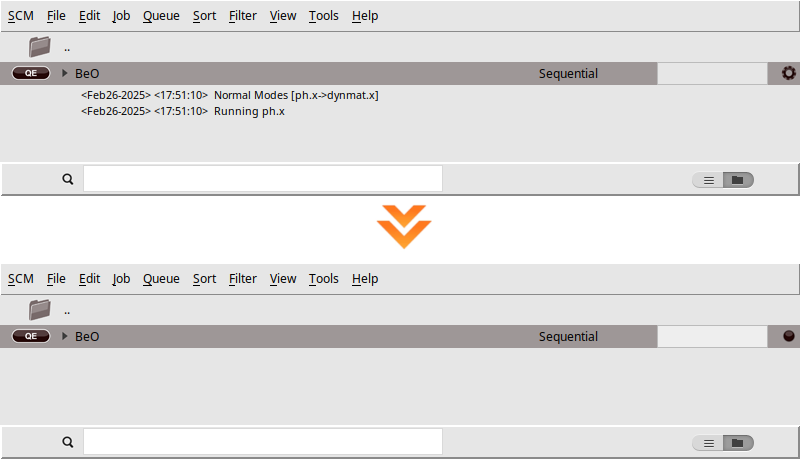
We are now ready to start the calculation
BeO)When you run a calculation, AMSjobs should come to the forefront, and your job will appear at the top of the list. On the right side, you’ll see the job running, indicated by a gear icon. While the simulation is in progress, the AMSjobs window will display the progress based on the logfile. Once the job is complete, the job status icon will change to a full circle to indicate that it is ready.
The AMSspectra module allows you to analyze both the IR and Raman spectra:
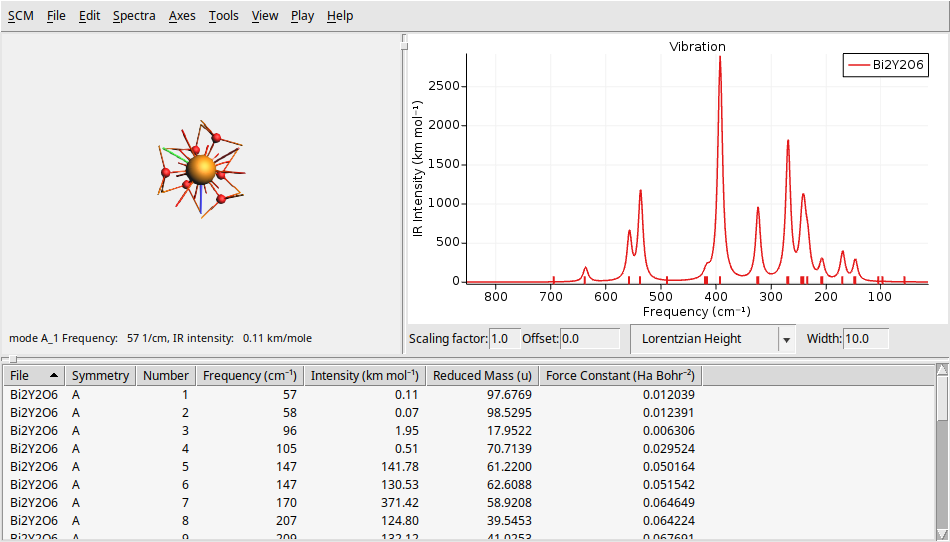
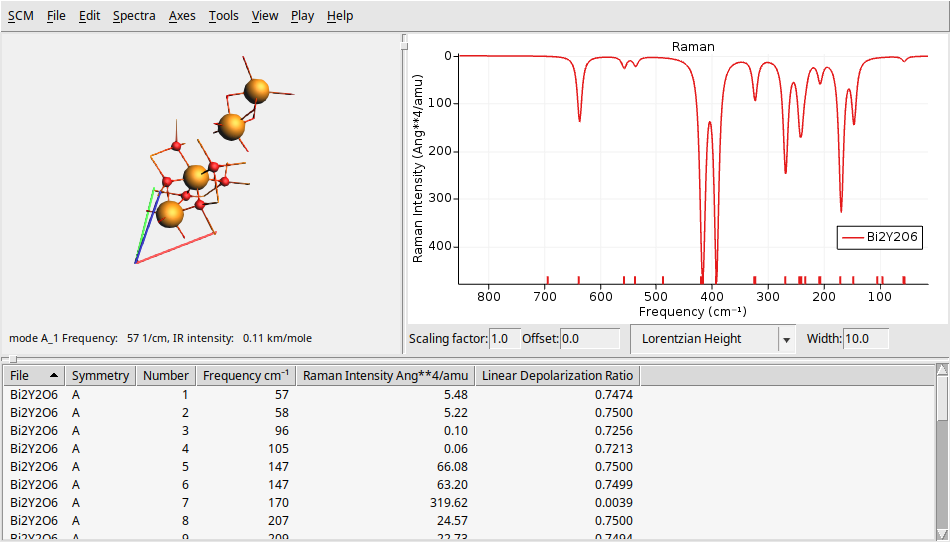
You should get something like this (note that the figure below uses a width of \(10.0\text{cm}^{-1}\), which may differ from your default value):
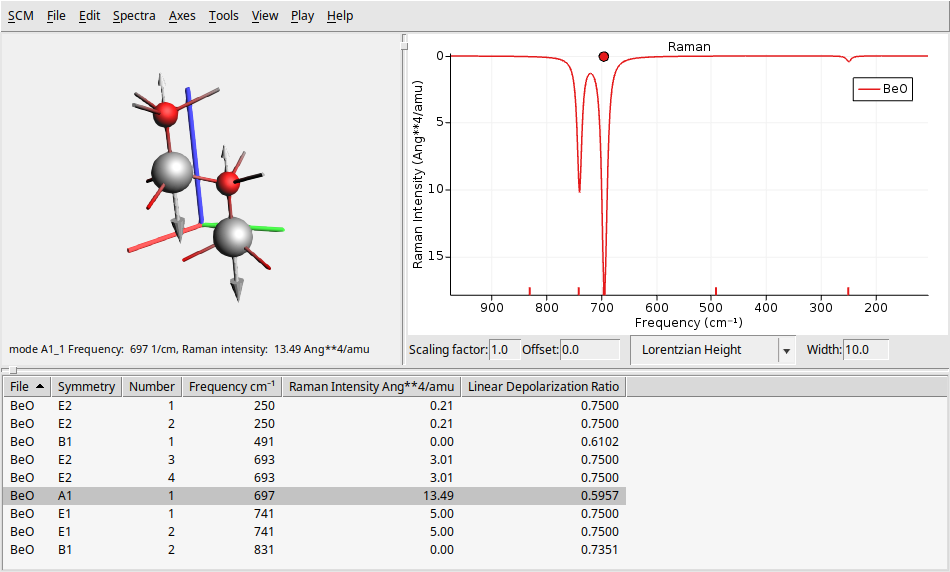
On the left side, there is a 3D representation of the BeO molecule, displaying the movement of atoms according to the selected normal mode. The right panel presents the calculated Raman spectrum. At the bottom, you will find a table that provides detailed information about each vibrational mode, including its symmetry, frequency, and Raman intensity.
Note
You can click on a vibrational mode in the bottom table or the Raman spectrum to see the atoms moving according to that mode’s vibrational frequency.
Note
To view the overlay arrows that indicate the displacement vectors for a specific vibrational mode, click on Play → Displacement Vectors. You can adjust the size of these arrows by using Ctrl + L to make them larger or Ctrl + K to make them smaller. Note that the figure above uses larger arrows than the default size.
Note
You can change several parameters related to the spectrum and its broadening using the text boxes at the bottom of the spectrum. You can modify the scaling factor and offset (useful for DFT functional correction factors), as well as the shape (such as Gaussian or Lorentzian) and the width.
To see the IR spectrum go to Spectra → Vibration. This will refresh the window to display the IR spectrum results in a layout similar to that of the Raman spectrum.
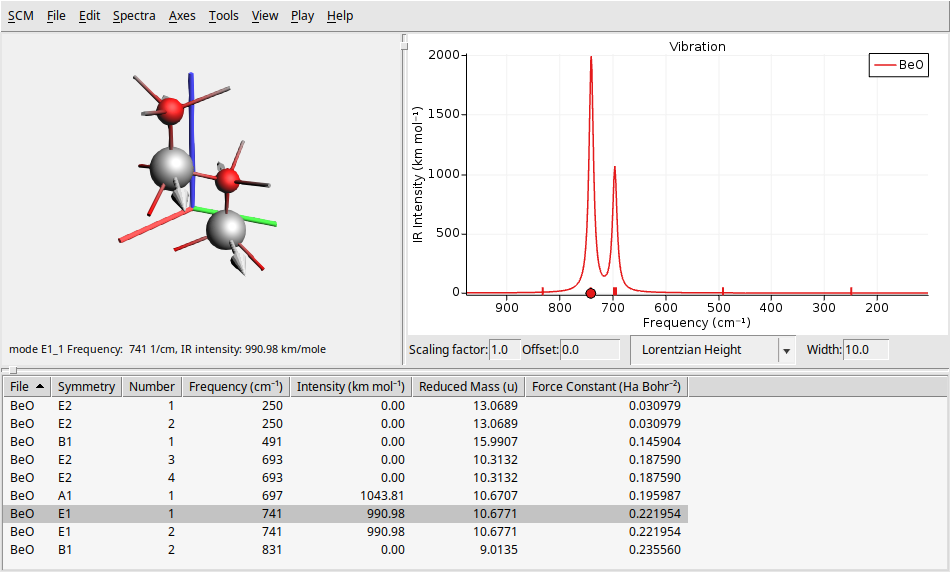
As we said before, our system BeO has a hexagonal structure with symmetry \(C_{6v}\) (6mm) symmetry. So, we can verify if the transitions found in our calculations are in agreement with the table of characters of the point group \(C_{6v}\)
\(C_{6v}\) |
E |
\(2C_6(z)\) |
\(2C_3(z)\) |
\(C_2(z)\) |
\(3σ_v\) |
\(3σ_d\) |
lin, rot |
quadrat |
|---|---|---|---|---|---|---|---|---|
\(A_1\) |
+1 |
+1 |
+1 |
+1 |
+1 |
+1 |
\(z\) |
\(x^2+y^2, z^2\) |
\(A_2\) |
+1 |
+1 |
+1 |
+1 |
-1 |
-1 |
\(R_z\) |
– |
\(B_1\) |
+1 |
-1 |
+1 |
-1 |
+1 |
-1 |
– |
– |
\(B_2\) |
+1 |
-1 |
+1 |
-1 |
-1 |
+1 |
– |
– |
\(E_1\) |
+2 |
+1 |
-1 |
-2 |
0 |
0 |
\((x, y) (R_x, R_y)\) |
\((xz, yz)\) |
\(E_2\) |
+2 |
-1 |
-1 |
+2 |
0 |
0 |
– |
\((x^2-y^2, xy)\) |
Note
Infrared selection rules:
If a vibration results in a change in the dipole moment, it is IR-active. These vibrational modes have the same symmetry as the \(x\), \(y\), and \(z\) axes.
Raman selection rules:
If a vibration results in a change in the polarizability, it is Raman-active. These vibrational modes have the same symmetry as quadratic functions like \(xy\), \(xz\), and \(x^2-y^2\).
From the characters table above, we can anticipate the IR and Raman activity of the BeO vibrational modes. This table indicates that modes with \(A_1\) and \(E_1\) symmetry should be IR-active, while modes with \(A_1\), \(E_1\), and \(E_2\) symmetry should be Raman-active. Notice this analysis is consistent with the results obtained from our calculations, as shown in the IR and Raman spectra plots above (see the columns symmetry and intensity at the bottom table).
Using Python (PLAMS)¶
The full PLAMS workflow is now maintained as a scripting example:
It contains both downloadable notebook/script files and the rendered step-by-step Python workflow.
See also:
Quantum ESPRESSO phonon band structure and DOS for BeO for the related phonon workflow.
Further Challenge¶
To further challenge your understanding, let’s calculate the IR and Raman spectra of a larger, and more complex system \(Bi_2Y_2O_6\) (10 atoms and 52 electrons). You can find theoretical results for comparison in The Computational Raman Database (mpid: mp-768221).
Y 0.000000029801245 -0.000000017205758 11.584484628486793
Y -0.000000000000000 -0.000000000000000 4.118754113258331
Bi 0.000000029801245 -0.000000017205758 14.817148949424018
Bi -0.000000029801246 0.000000017205756 7.351418334652482
O 1.327040764604285 -1.891211026034423 8.229234902990678
O 1.653083765594469 -0.170635326573514 5.740657998219140
O -0.974316410415964 -1.346294872078748 5.740657998219141
O -2.301357234622741 -0.203645500808650 8.229234902990676
O 0.974316380614717 2.094856578460344 8.229234902990676
O -0.678767355178506 1.516930198652259 5.740657998219140
VEC1 2.980 1.721 4.977
VEC2 -2.980 1.721 4.977
VEC3 0.000 -3.441 4.977