Accurate XC functionals¶
This tutorial will showcase some of the accurate XC functionals included in ADF that can be used for barrier heights, reaction energies, and non-covalent interactions: the fast r2SCAN-3c composite method, the accurate sigma-functional (σ-functional), a general-purpose double hybrid B2GPPLYP-D3BJ, and an opposite-spin-only double hybrid rev-DOD-PBEP86-D4. The section on XC functionals in the ADF manual provides more technical details on these functionals.
We will be calculating the barrier height for a (water-catalyzed) proton-transfer reaction. This is one of the reactions in the WCPT18 [1] subset of the GMTKN55 database [2]. These types of databases are often used when benchmarking the performance of various XC functionals. In literature one can also find recommendations for which XC functional to use in which situation.
While ADF contains many XC functionals, a few of them are highlighted here: The r2SCAN-3c composite method and the σ-functional were chosen as they include specific parametrizations for STO basis sets. B2GPPLYP-D3BJ and rev-DOD-PBEP86-D4 were chosen because of the well-documented precision of double hybrid functionals.
r2SCAN-3c¶
The r2SCAN-3c composite method [3] uses the \(r^2\) SCAN (r2SCAN) exchange-correlation functional, in combination with a tailor-made all electron polarized basis set (mTZ2P), the semiclassical London dispersion correction (D4), and a geometrical counterpoise (gCP) correction. The STO-optimized r2SCAN-3c outperforms many conventional hybrid/QZ approaches across different applications, at a fraction of the cost. The r2SCAN-3c method in ADF supports both single point calculations and geometry optimizations.
Sigma-functional¶
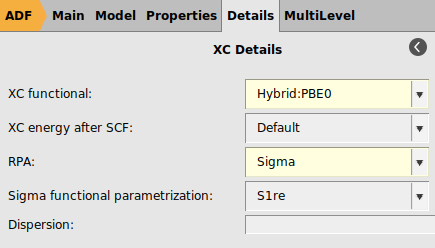
σ-functionals belong to the class of Kohn–Sham (KS) correlation functionals based on the adiabatic-connection fluctuation-dissipation theorem and are closely related to the random phase approximation (RPA). In Ref. [4] it was concluded that σ-functionals are highly promising alternatives to established density functionals, including double hybrids. ADF features a σ-functional implementation which is specifically parametrized for STO-type basis sets [4]. In particular, for the TZ3P basis one should use the S1re parametrization of the σ-functional. Note that σ-functionals in ADF can only be used in single point calculations.
Note
For QZ4P or QZ6P basis sets, one should use the S1 parametrization of the σ-functional instead.
Double hybrids¶
Double-hybrid functionals usually yield considerably better energies than (meta-)GGA and (meta-)hybrid functionals for (main group) thermochemistry and kinetics, transition metal chemistry and non-covalent interactions. B2GP-PLYP [5] is a general-purpose double hybrid, aimed at predicting a range of properties across different systems. Rev-DOD-PBEP86-D4 [6] is an opposite-spin-only double hybrid. Opposite-spin only double hybrid functionals are recommended for large systems (50-100 atoms and larger) since they are computationally more efficient than the other double hybrid functionals which also include the same-spin contribution.
Note that double hybrids converge much slower to the complete basis set limit than simpler DFT methods. In ADF, double hybrids can only be used in single point calculations.
Tip
For specific properties, the optimal choice of double hybrid functional may differ. Consult the manual for more details.
Calculation settings¶
We start by making an input file for a single point energy calculation. Instructions are given below for each of the XC functionals. The input coordinates will be specified later, so we can leave the molecule editor empty for now.
When saving the file, AMSinput will automatically adjust the Basis Set to mTZ2P, the Frozen Core to None and the Numerical Quality to Good, as required by r2SCAN-3c.
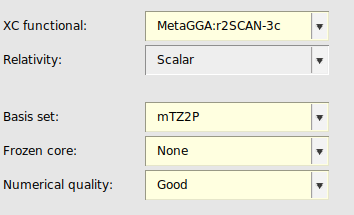
 next to the XC Functional
next to the XC Functional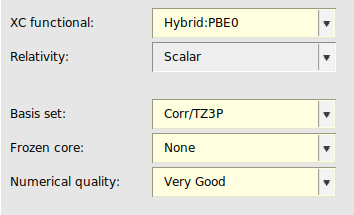
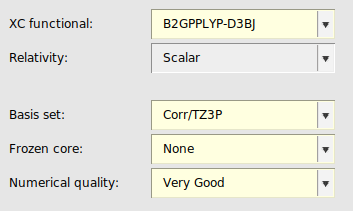
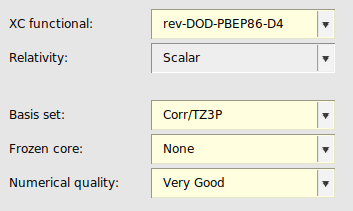
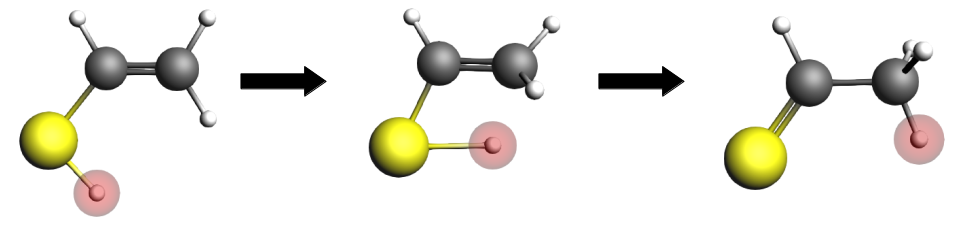
IS9 → TS9 → FS9¶
We will now perform single point calculations for the initial state (IS9, the enol), the transition state (TS9) and the final state (FS9, the ketone).
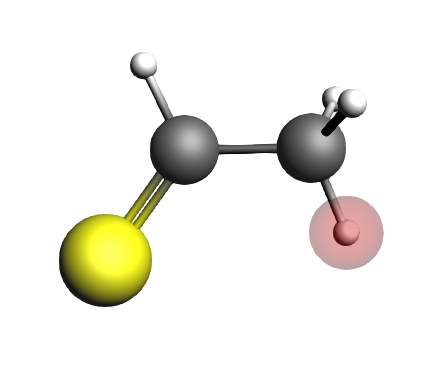
C 0.000000 0.759038 0.000000
H -0.781174 1.509717 0.000000
C 1.280382 1.112661 0.000000
H 2.084515 0.388850 0.000000
H 1.555670 2.157995 0.000000
H 0.468551 -1.541748 0.000000
S -0.688116 -0.859063 0.000000
Run the calculation:
We can now repeat these steps for the transition state:
C 0.468987 0.636874 0.053256
H 0.585413 1.716908 0.037321
C 1.515535 -0.285885 -0.030947
H 1.542575 -1.062299 0.729004
H 0.301002 -1.075601 -0.411970
H 2.498769 -0.002264 -0.395534
S -1.052181 -0.105168 -0.005792
And for the final state:
C 0.000000 0.617005 0.000000
H 0.517521 1.577301 0.000000
C -1.485898 0.714108 0.000000
H -1.957093 -0.265537 0.000000
H -1.819218 1.282178 0.874496
H -1.819218 1.282178 -0.874496
S 0.874587 -0.741425 0.000000
The output from these calculations can be used to compute the reaction energies and reaction barriers. The results have been summarized in the table below:
barrier height |
reaction energy |
|
|---|---|---|
Ref [1] |
58.80 |
-2.44 |
r2-SCAN-3c |
54.1 |
-3.7 |
σ-functional (S1re) |
57.3 |
-2.2 |
B2GPPLYP-D3BJ |
59.0 |
-2.1 |
rev-DOD-PBEP86-D4 |
59.8 |
-1.9 |
Note
The results for 1 specific molecule are insufficient to determine the general accuracy of an XC functional. Typically, larger benchmark studies are performed on many molecules when comparing XC functionals.
QZ6P basis set¶
Since double hybrids converge much slower to the complete basis set limit than simpler DFT methods, we include here an overview of the settings for calculations with the larger QZ6P basis set. The r2SCAN-3c composite method has only been defined for the mTZP basis set, and will therefore be omitted here.
Note
The use of QZ6P basis sets with σ-functionals or double hybrids is only recommended for expert ADF users as it may be more complicated to attain numerical stability. See also the recommendations for MBPT calculations in the ADF manual.
Note
Because the S1re parametrization of the σ-functional is only valid for TZ3P basis sets, one has to select the S1 parametrization when using a QZ6P basis.
next to the XC Functional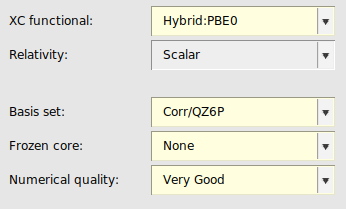
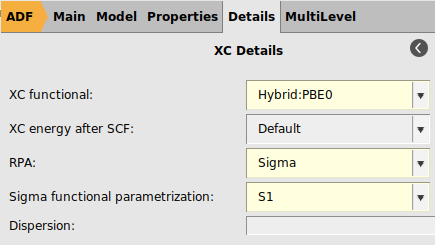
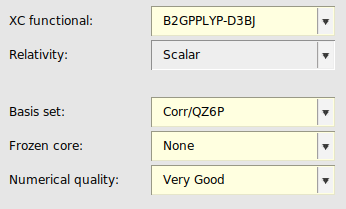
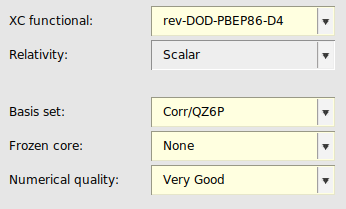
The results for the QZ6P calculations have been summarized below:
barrier height |
reaction energy |
|
|---|---|---|
Ref [1] |
58.80 |
-2.44 |
σ-functional (S1) |
59.5 |
-1.3 |
B2GPPLYP-D3BJ |
58.7 |
-2.5 |
rev-DOD-PBEP86-D4 |
59.6 |
-2.3 |
We remark again that the results for 1 individual calculation, or 1 individual property, cannot be used to make conclusions on the accuracy of the functional as a whole. One would typically perform calculations on a larger number of molecules to determine which functional is most reliable for a specific application. If computational resources are limiting, one may also consult literature to find a suitable functional based on prior analyses.