Modeling phosphorescent lifetimes of OLED emitters¶
In organic light-emitting diodes (OLEDs), phosphorescent dyes can be advantageous compared to fluorescent dyes because of the increase in the maximum theoretical efficiency from 25% to 100%. Both can me modeled with AMS, but to model the spin-forbidden phosphorescence (which involves a transition from the triplet state to the singlet ground state, T1 → S0 ) spin-orbit couplings must be included in TDDFT calculation (SOC-TDDFT). This guided example explains how to use ADF to calculate phophorescent lifetimes using SOC-TDDFT, in the case of famous OLED emitter, the 5d transition metal complex Ir(ppy)3 .
Example input and output files¶
While this example is self-contained, it is still helpful to download the starting geometry.
Note that this starting geometry is specifically the Λ-fac-Ir(ppy)3 isomer, though other isomers can also be investigated in the same way.
Note
This example may take quite some time, we recommend running the calculations on a HPC cluster if possible.
Essentially the procedure for predicting phosphorescent lifetimes consists of two steps:
Geometry optimization of the lowest triplet state (T1)
SOC-TDDFT calculation at the optimized T1 geometry
In ADF there are two ways to include spin-orbit couplings in a calculation: you can do a fully self-consistent variational SOC-TDDFT calculation, or do a perturbative SOC (pSOC) calculation. A pSOC-TDDFT calculation offers insight in the underlying singlet and triplet contributions to the excitations, which can aid with designing ligands which yield better emission properties. Similarly, insight in singlet and triplet contributions may also be achieved by performing a full SOC calculation employing a scalar-relativistic fragment of the full molecule, as explained further below.
Optimization of the lowest triplet state (T1)¶
Since the T1 state is the lowest in the triplet manifold, the T1 geometry can be optimized just as a regular ground state, but with triplet spin multiplicity. Usually the triplet state can break the symmetry of the singlet ground state, so it is recommended to switch off symmetry (NoSym). See also the paper by Gonzalez-Vazquez et al. [1] where the importance of relaxing the T1 state is discussed.
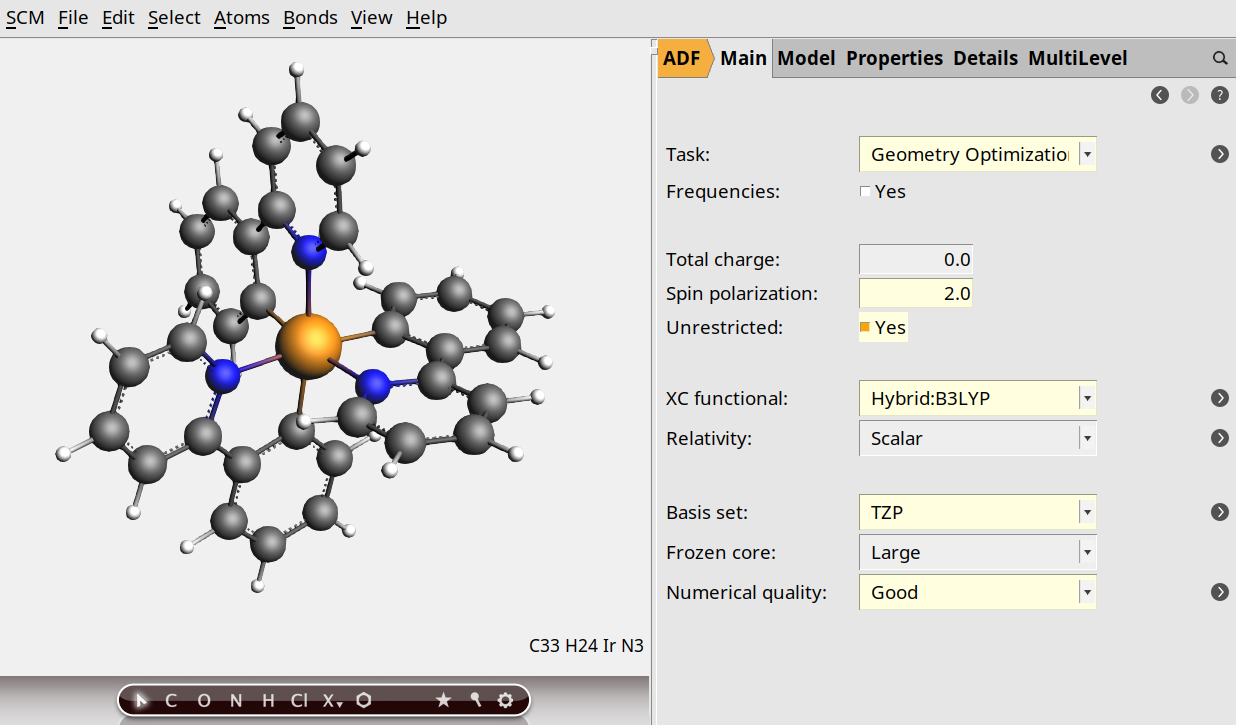
In the example we optimize the triplet state with the B3LYP functional, a TZP basis set, scalar ZORA for relativistic effects and we set the numerical quality to Good. Just follow these steps in the GUI:
Note
You may ignore warnings about using hybrid functionals and frozen core in this case since we are not interested in the properties of core electrons
Tip
Want a faster geometry optimization? You can use a lower level of theory such as PBE-D3/DZP which can be orders of magnitude faster and only use the higher level of theory for the phosphorescence calculations
Relativistic spin-orbit coupling TDDFT calculation¶
To calculate the phosphorescent lifetime from the T1 state, a SOC-TDDFT calculation is performed on the molecule in the T1 optimized geometry. This method converges the singlet state first, and then uses response theory (TDDFT) to model the transition to and from the T1 state.
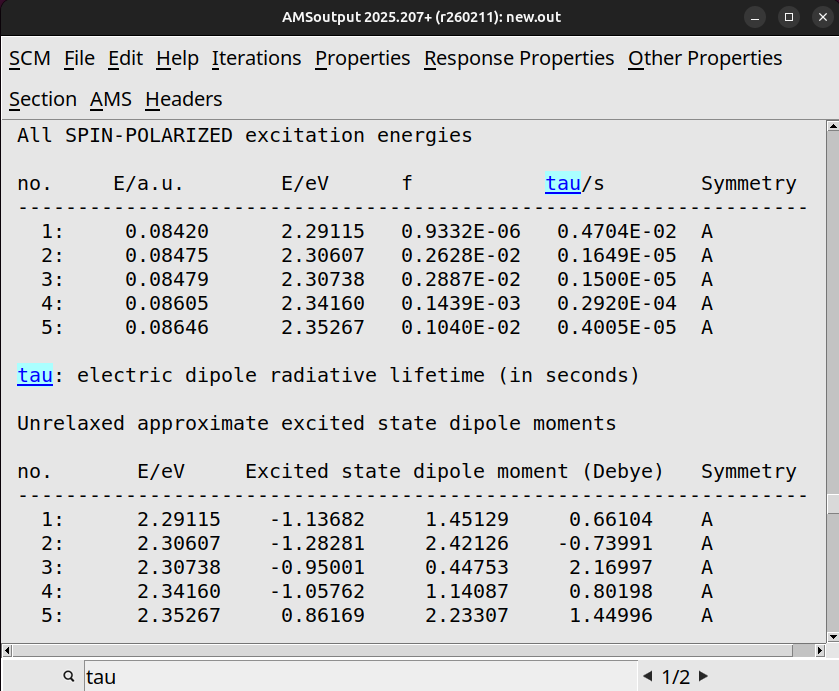
Typically the lowest 3 states of the spin-orbit coupling TDDFT calculation are important which resemble the 3 states in a triplet state. There will be a small energy difference between the three states because of spin-orbit coupling, the so called zero-field splitting (ZFS). The oscillator strengths and radiative lifetimes \(\tau\) for each transition are printed in the output, and are related according to:
Where \(\hbar\) is the reduced Planck constant, \(\Delta E\) is the energy difference between the two states, \(m_\text{e}\) is the electron mass, \(c\) is the speed of light, \(\alpha\) is the fine structure constant, and \(f\) is the oscillator strength of the transition.
To set up a SOC-TDDFT for your own phosphorescent dye you can first load the optimized triplet structure obtaine in the previous step and follow these instructions in the GUI:
Save then run the calculation.
Output: phosphorescent lifetimes¶
The ouput file may be opened either with your favorite text editor or with the ADFGUI.
If you open it with the GUI, it will take you to the output browser. You can search for ‘tau’ to jump straight to the phosphorescent lifetimes.
If you scroll up from the radiative lifetimes you will find the major MO → transitions.
A cheaper alternative (ca. 5 times faster) to full spin-orbit coupling calculations is the approximate, perturbative SOC scheme. This seems a reasonable approximation for phosphorescence radiative lifetime calculations, as for instance found in this highlight on OLED phosphorescence. [6]
Perturbative SOC: spin-orbit coupling matrix (SOCME)¶
The pSOC method does not include spin-orbit coupling in the ground state at all, instead they are included purely in the response calculation using perturbation theory. This leads to considerable computational savings.
Setting up the calculation is simple (see instruction below). One difference is the fact that more excitations should be converged, since they are used in the perturbative expansion. Increasing the number of converged states leads to better convergence, at the price of a higher computational cost.
You may then save and run your calculation. The results are presented the same as before, though be aware that in this case singlets, triplets, and perturbative states are calculated sequentially therefore you will have to scroll down and right before the lifetimes you can find the spin-orbit couplings matrix elements.
These SOCMEs are important parameters for designing phosphorescent OLED emitters: optimize internal conversion to T1 from the singlet exciton and minimize non-radiative decay. Likewise, SOCMEs are useful figure of merits to design better TADF (thermally activated delayed fluorescence) emitters. [5]
Accuracy considerations of the phosphorescent lifetime predictions¶
The use of the XC functional is relevant for the accuracy of the results. In this example the B3LYP functional was used. One can also use other hybrids by chosing the appropriate fucntional in the GUI (main panel) or setting the appropriate functional in the XC block in your input. Hybrid functionals such as B3LYP seem to give quite good results, however, they will take more CPU time compared to pure functionals. You may even use frozen cores, although this is not strictly correct for hybirds.
We find good agreement between calculated and experimental radiative lifetimes for various phosphorescent transition metal complexes when we use the following settings:
Strict presets: ExactDensity, FullSCF, gradients 1e-4, SCF 1e-6, Becke ‘Good’ grid, DEPENDENCY bas=1e-3
All-electron DZP basis (defaults to TZP for transition metals)
Optimize T1 with reduced symmetry, or NoSym
B3LYP functional
20 excitations for the SR-TDDFT calculation, 4 for the STCONTRIB SOC-TDDFT calculation
Further accuracy considerations may be to employ a larger basis set (TZ2P) and to include continuum solvation with COSMO. Solvation effects may especially improve the accuracy of ZFS and lifetime calculations [2] .
In a study by Younker and Dobbs, a good correlation is found between calculated and observed phosphorescent rates of Ir(III) complexes when using a pSOC-TDDFT approach with B3LYP on the BP86-optimized singlet ground state. [6]