Resin Blend from Molecular Dynamics¶
This tutorial will teach you how to:
build a small polyisoprene/H-DCPD resin blend based on Ref. [1];
equilibrate a pseudo-2D bulk polymer cell with UFF and ReaxFF;
monitor the density during annealing;
analyze the miscibility of the resin.
Introduction¶
Rissanou et al. [1] investigated hydrogenated dicyclopentadiene (H-DCPD) resin blended with polyisoprene (PI) and SBR. For the PI system at high resin loading, the paper used 100 PI chains with 232 H-DCPD resin molecules. This tutorial scales that composition down to 20 PI chains and 46 H-DCPD molecules, which gives a system of about 10,000 atoms.
The goal is not to reproduce the statistical quality of the production paper. Instead, the tutorial reproduces the workflow: packing the blend, compressing the low-density starting structure, annealing the material, and analyzing resin-rich structure.
You can run this tutorial with Python from the companion notebook
or the Python workflow.
Downloads¶
Use these structures as inputs for the tutorial:
Note
Refer to Building Polymers to build the PI chain. You can define the cis-1,4-isoprene molecule, define the anchors, and generate a 30-mer.
Part A: Build the Blend¶
Create the packed PI/H-DCPD structure¶
0.05 g/cm 3PI_30mer.xyz20 and Region to polymerHDCPD_trimer.xyz46 and Region to resinNote
The paper used 100 PI chains and 232 resin molecules. The 20:46 system used here preserves the same approximate composition while keeping the tutorial small enough for a local workstation.
Part B: Equilibrate the Polymer Bulk¶
Run a UFF geometry relaxation¶
UFF_InitialRelaxPre-shrink to a pseudo-2D bulk cell¶
We will intentionally create a pseudo-2D periodic bulk cell with
a' = b' = λc' (λ > 1). This makes the later resin-density map easier to
interpret while still using 3D periodic boundary conditions.
The target volume V as a function of the initial volume, initial density, and target density is therefore:
We can then deduce the target cell dimensions as a function of the c-scaling factor λ:
We will use λ = 1.5 and the final target density ρ = 0.9 g/cm 3.
Tip
If you open UFF_InitialRelax with AMSinput, you can easily read the cell volume and
density from Properties → Properties (Including Estimated).
UFF_InitialRelax and Update Molecule200000.25 fs10005 K5 K, and the Damping Constant to 10 fs53 53 35UFF_Shrink
The calculation takes about 20 minutes on an Apple M2 with 16 GB of memory.
Run a ReaxFF relaxation¶
UFF_Shrink in AMSinputdispersion/CHONSSi-lg.ffReaxFF_InitialRelaxNote
If the geometry optimization reaches a plateau with small gradients, you can request an early stop in AMSjobs by selecting Jobs → Request Early Stop. Tight convergence is not required here because an MD simulation will be run immediately afterward. However, do not skip this step: the forces should still be minimized before starting the MD simulation to help prevent the molecule from decomposing.
Part C: Anneal and Produce a Trajectory¶
Run ReaxFF annealing at 413 K¶
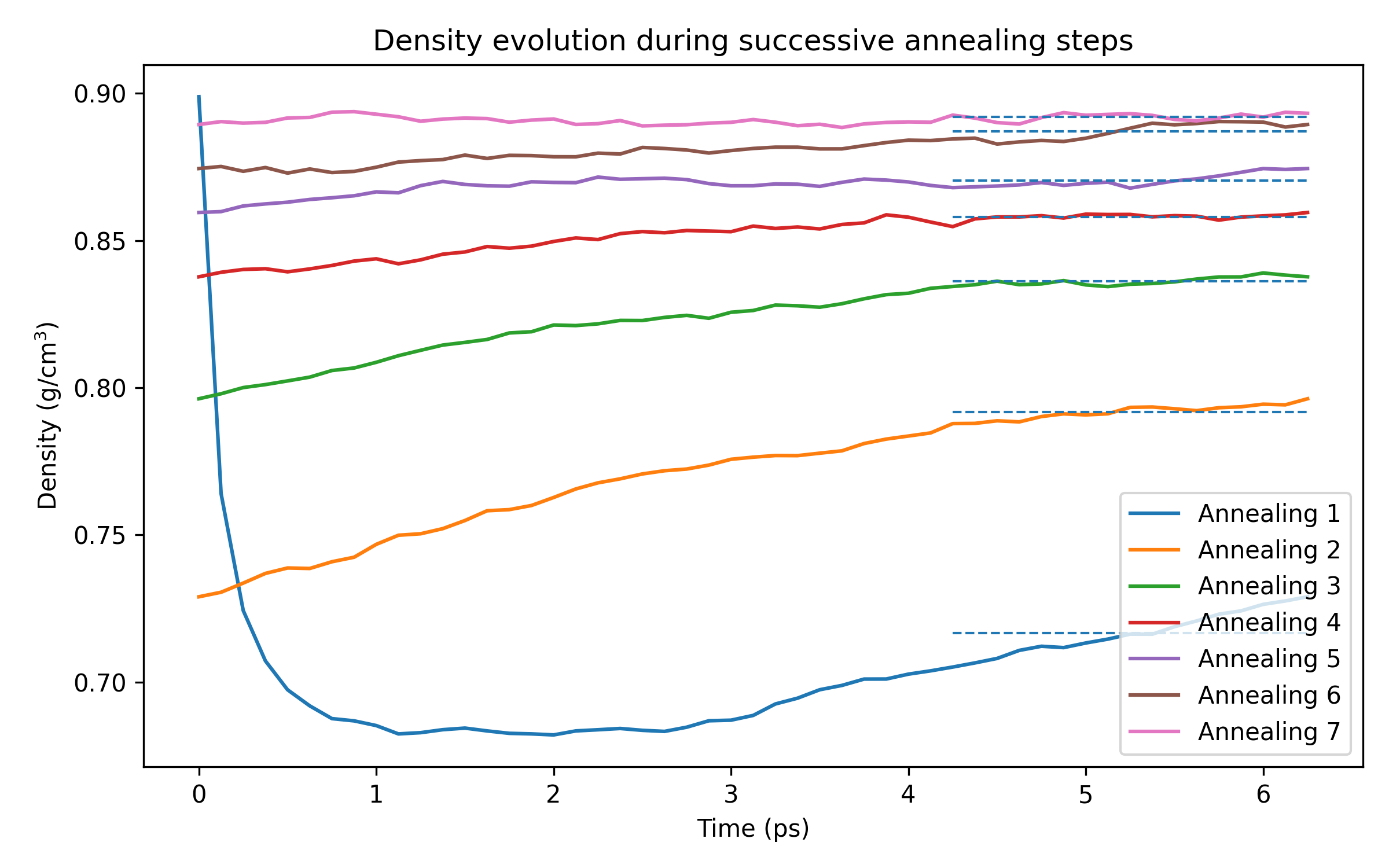
ReaxFF_InitialRelax in AMSinputdispersion/CHONSSi-lg.ff force field25000500413 K298.15 413 298.15 K, Duration to 12500 12500 and Tau to 100 fs101325 Pa, and Tau to 500 fsReaxFF_Anneal_1 and runRepeat the procedure from the final structure (update the molecule and save) until the last-third
average density changes by less than about 0.5 percent.
Tip
To get the density, open the job in AMSmovie, Graph → Add Graph, plot the density with MD Properties → Density, and save it to a file with Graph → Save as XY. Then you can get the last-third average from a terminal (from the same folder as the data file):
awk 'NR==FNR && FNR>1 {total++; next} FNR>1 {i++; if (i >= int(2*total/3)+1) {sum+=$2; n++}} END {print sum/n}' rho_1.xy rho_1.xy
Annealing Step |
Initial Density |
Final Density |
Last Third Average Density |
% Change |
|---|---|---|---|---|
1 |
0.8988 |
0.7290 |
0.7168 |
_ |
2 |
0.7290 |
0.7962 |
0.7917 |
9.46 |
3 |
0.7962 |
0.8377 |
0.8362 |
5.32 |
4 |
0.8377 |
0.8595 |
0.8580 |
2.54 |
5 |
0.8595 |
0.8744 |
0.8704 |
1.42 |
6 |
0.8744 |
0.8893 |
0.8871 |
1.88 |
7 |
0.8894 |
0.8932 |
0.8920 |
0.55 |
Run a production trajectory¶
dispersion/CHONSSi-lg.ff50000100413 K413 K with Tau 100 fsReaxFF_Production and runPart D: Analyze the Trajectory¶
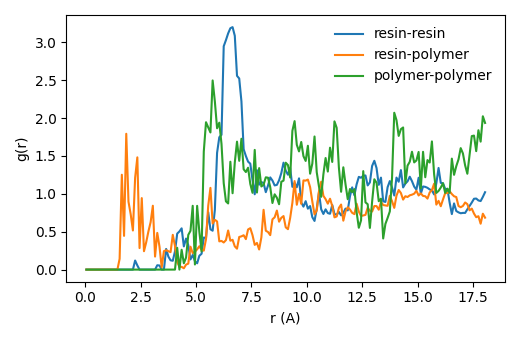
Radial distribution function¶
To understand the miscibility of the resin in the polymer matrix, we compute and report the
resin-resin and resin-polymer center-of-mass RDFs. You can use this
PLAMS script and simply modify the path so
the trajectory points to the ams.rkf of the final production run.
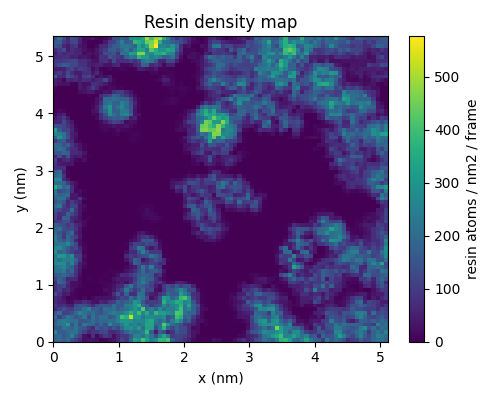
Density map¶
The miscibility can also be appreciated by plotting the 2D density map of the resin atoms.
You can use this PLAMS script to generate
and plot the density map.
Note
To get better statistics, you should run a longer production trajectory, increase the sampling frequency, or perform the simulations with a larger system size.
Summary¶
You built a small PI/H-DCPD blend, compressed it into a pseudo-2D bulk cell, annealed it with ReaxFF, and inspected the miscibility of the resin.