Usage of pdb2adf¶
Short description¶
The program works interactively, and should be straightforwardly to use. However, for some of the stages in the output a short description is given below.
P D B 2 A D F - program
version 2005.01
Written by: Marcel Swart, 2005
This program uses AMBER parameter files
see: http://amber.scripps.edu
Do you want a logfile to be written (Y/n) ?
This option exists to create a logfile of what pdb2adf does. However, it should normally be used only for debugging purposes.
Ignoring atom on line:
ATOM 974 OH LYS A 128 -10.073 42.775 15.690 1.00 38.79 5AZU1065
This is a warning that the atom on that particular line is ignored, should normally occur only few times (less than ten). Depends also on how well the PDB file follows the PDB format rules.
Data Processed:
Nat: 2519
Nmol: 196
NChains: 1
Information about what has been read on the PDB file: the total number of atoms (Nat), number of molecules/residues (Nmol) and number of protein chains (Nchains).
Please wait, making connection tables
At this point, the connections between the atoms are being made by looking at atom distances. It may take a while, depending on the size of the system.
Do you want to make separate files for each chain (Y/n) ?
You have the option to make different inputfiles for different protein chains, but you can also make one inputfile for all of them together.
Found the following terminal amino acid residues : (C-term) 128 (N-term) 1
Do you want to use these as terminal residues (Y/n) ?
Info is given about the C- and N-terminal residue of each chain. Reported for making sure they are chosen correctly. Note, if the C- and N-terminal residues are connected (rarely the case probably), enter N here.
Multiple AMBER options for HIS :
0 Decide every time differently
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Suggested option: 0
For a number of residues (His, Glu, Asp, Lys and Cys) there is more than one option available in the AMBER95 force field, depending on the protonation state (His, Glu, Asp and Lys) or the existence of a sulphur bridge/connection to a metal atom (Cys). The default is to choose a different option for the His and Cys residues, and use one option for Glu, Asp and Lys (fully charged). However, if wanted you can make a choice for all residues.
Multiple AMBER options for CYS 3 ( 3) :
1 CYS Cysteine (SH)
2 CYM Deprotonated Cysteine (S-)
3 CYX Cystine (S-S bridge)
Connections and Nearest Atoms for SG CYS 3 SG ( P2A # 41 PDB# 20 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.82 38 19 CB CYS 3 CB 1 3.79 2382 980 O HOH 151 O
2 2.02 461 193 SG CYS 26 SG 2 3.80 22 0 HC GLN 2
3 4.04 2391 983 O HOH 154 O
4 4.15 509 206 O GLN 28 O
5 4.18 522 0 HA PHE 29
Suggestion: 3
The options for Cys3 are given, with information about the atoms bonded to the SG sulphur atom (on the left), as well as the closest five non-bonded atoms (on the right). This information may help you decide which choice to make for this particular residue. Also given (on the bottom) is the suggested choice, which is based, in this case, on the presence of a sulphur bridge.
Multiple AMBER options for HIS 46 ( 46) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 46 ND1 ( P2A # 844 PDB# 347 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.37 843 346 CG HIS 46 CG 1 2.62 2166 0 H1 MET 121
2 1.33 846 349 CE HIS 46 CE1 2 3.23 2080 863 ND HIS 117 ND1
3 2.04 2318 959 CU CU 130 CU 3 HB 3.33 2163 900 S MET 121 SD
4 3.40 2164 901 CT MET 121 CE
5 3.57 2082 865 CE HIS 117 CE1
Connections and Nearest Atoms for NE HIS 46 NE2 ( P2A # 848 PDB# 350 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.32 846 349 CE HIS 46 CE1 1 HB 2.70 162 67 O ASN 10 O
2 1.37 850 348 CD HIS 46 CD2 2 2.83 814 0 H1 MET 44
3 3.23 2166 0 H1 MET 121
4 3.52 822 332 O MET 44 O
5 3.74 813 334 CT MET 44 CG
Suggestion: 2
For His residues, the information is given for both the delta- and the epsilon nitrogen atoms. Also indicated (by HB) is the presence of a hydrogen bond with another atom. The definition used here is that two atoms are hydrogen bonded if they are both non-carbon/non-hydrogen atoms, and the distance between them is less than the sum of the van der Waals radii of the atoms. It is a simple definition, but seems to be effective. In this case, as the N(delta) is bonded to copper, the proton should be attached to the N(epsilon).
Making choice for which molecules should be QM, which MM
Now we come to the part where the division in the QM and MM systems is made.
Residues belonging to chain 0
Option Molecule Option Molecule Option Molecule Option Molecule Option Molecule
1: ALA 1 28: GLN 28 55: ASP 55 82: ALA 82 109: ALA 109
2: GLN 2 29: PHE 29 56: LYS 56 83: HIS 83 110: TYR 110
etc
All molecules/residues belonging to chain 0 are given, with an option number.
Give option number of molecules to be put in QM region (or 'c' to continue):
Note: by specifying a negative number a molecule is removed from the QM region
Here you are asked to enter the option numbers of the residues you want to put in the QM system.
Putting GLY 45 in QM region
Putting HIS 46 in QM region
In this case, Gly45 and His46 have been put in the QM system.
Make a choice for the QM/MM treatment of GLY 45
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 2
Give choice:
A choice should be made for where to cut-off the QM system. Normally this is done at the C(alpha) position, and you should simply choose the Suggestion.
Solvent molecules (SOL/HOH) belonging to this chain:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
61 62 63 64 65 66
Give the number of the molecule to be put in QM region (or 'c' to continue):
Also water molecules can be put in the QM system.
Box Shape options:
1 Spherical box
2 Cubic box
Make a choice:
Type of box to be used.
Maximum atomic distance (Angs) from center 25.62
Give boxsize (def.: 28.62 Angs)
Size of box to be used to put a layer of solvent molecules around the system. Max. dist. is the maximal distance of any protein atom from the center of mass of the protein. Usually you should choose a boxsize at least 6 Angstrom larger (so at least two solvent molecules are surrounding the system).
...
Using BOXSIZE value of 30.0000
Adding atoms for box 1 Added (Box): 0 (Total): 0 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 2 Added (Box): 9 (Total): 9 Excl. (1): 639 Excl. (2): 0
Adding atoms for box 63 Added (Box): 3 (Total): 7635 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 64 Added (Box): 0 (Total): 7635 Excl. (1): 648 Excl. (2): 0
Writing inputfile for chain 1
A total amount of 7635 atoms (2545 water molecules) has been added.
Inputfile(s) written, everything processed, work has been done.
Thank you for using the PDB2ADF program.
================================
Normal ending of PDB2ADF program
================================
ADF inputfile(s) have been written, the PDB-file has been processes. Everything is done.
An example on protein structure¶
The idea of this example is to make an adf-input file using a PDB of an azurin (1DYZ.pdb). The result of this example should be that in the adf-input file the active site of azurin (Figure 1) is in the QM part, and the rest of the protein is in the MM part, and that the solvent water is added (in a box), which is also in the MM part.
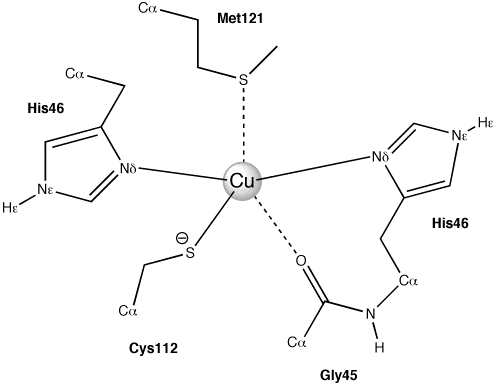
Figure 1: the active site of azurin
Usage of pdb2adf¶
The program works interactively. Given below in bold are the parts that the user has to type. In cases where the user agrees with the suggestion given by the program, the user can press the Enter key indicated with Enter.
P D B 2 A D F - program
version 2005.01
Written by: Marcel Swart, 2005
This program uses AMBER parameter files
see: http://amber.scripps.edu
Do you want a logfile to be written (Y/n) ?
Enter
Please give name of PDB-file
1DYZ.pdb
read fragments
Data Processed:
Nat: 2519
Nmol: 196
NChains: 1
Please wait, making connection tables
Now finding nearby atoms
Assigning chain ID to all residues
Completing residues for which only option is available
Found the following terminal amino acid residues : (C-term) 129 (N-term) 1
Do you want to use these as terminal residues (Y/n) ?
Enter
Refinding nearby atoms (including atoms added in residue completion)
Multiple AMBER options for HIS :
0 Decide every time differently
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Suggested option: 0
Enter
Using 0: Decide every time differently
Multiple AMBER options for GLU :
0 Decide every time differently
1 GLU Glutamic acid (COO-)
2 GLH Neutral Glutamic acid (COOH)
Suggested option: 1
Enter
Using 17 GLU Glutamic acid (COO-)
Multiple AMBER options for ASP :
0 Decide every time differently
1 ASP Aspartic acid (COO-)
2 ASH Neutral Aspartatic acid (COOH)
Suggested option: 1
Enter
Using 18 ASP Aspartic acid (COO-)
Multiple AMBER options for LYS :
0 Decide every time differently
1 LYS Charged Lysine (NH3+)
2 LYN Neutral Lysine (NH2)
Suggested option: 1
Enter
Using 19 LYS Charged Lysine (NH3+)
Multiple AMBER options for CYS :
0 Decide every time differently
1 CYS Cysteine (SH)
2 CYM Deprotonated Cysteine (S-)
3 CYX Cystine (S-S bridge)
Suggested option: 0
Enter
Using 0: Decide every time differently
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Making Choices for Chain 0
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Multiple AMBER options for CYS 3 ( 3) :
1 CYS Cysteine (SH)
2 CYM Deprotonated Cysteine (S-)
3 CYX Cystine (S-S bridge)
Connections and Nearest Atoms for SG CYS 3 SG ( P2A # 41 PDB# 20 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.82 38 19 CB CYS 3 CB 1 3.79 2382 980 O HOH 151 O
2 2.02 461 193 SG CYS 26 SG 2 3.80 22 0 HC GLN 2
3 4.04 2391 983 O HOH 154 O
4 4.15 509 206 O GLN 28 O
5 4.18 522 0 HA PHE 29
Suggestion: 3
Enter
Multiple AMBER options for CYS 26 ( 26) :
1 CYS Cysteine (SH)
2 CYM Deprotonated Cysteine (S-)
3 CYX Cystine (S-S bridge)
Connections and Nearest Atoms for SG CYS 26 SG ( P2A # 461 PDB# 193 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.82 458 192 CB CYS 26 CB 1 3.41 522 0 HA PHE 29
2 2.02 41 20 SG CYS 3 SG 2 3.43 411 168 O ASP 23 O
3 3.60 2322 960 O HOH 131 O
4 3.91 403 169 CB ASP 23 CB
5 4.15 387 0 HC VAL 22
Suggestion: 3
Enter
Multiple AMBER options for HIS 32 ( 32) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 32 ND1 ( P2A # 581 PDB# 244 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.39 580 243 CG HIS 32 CG 1 3.41 545 0 HC THR 30
2 1.33 583 246 CE HIS 32 CE1 2 3.43 76 33 O ALA 5 O
3 3.58 90 40 OH THR 6 OG1
4 3.99 91 0 HO THR 6
5 4.17 68 0 H ALA 5
Connections and Nearest Atoms for NE HIS 32 NE2 ( P2A # 585 PDB# 247 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.31 583 246 CE HIS 32 CE1 1 2.86 544 0 HC THR 30
2 1.37 587 245 CD HIS 32 CD2 2 3.00 545 0 HC THR 30
3 3.14 1677 0 HO SER 94
4 3.42 542 229 CT THR 30 CG2
5 3.65 1676 688 OH SER 94 OG
Suggestion: 1
3
Multiple AMBER options for HIS 35 ( 35) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 35 ND1 ( P2A # 649 PDB# 271 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.38 648 270 CG HIS 35 CG 1 2.46 682 0 H GLY 37
2 1.32 651 273 CE HIS 35 CE1 2 2.69 1604 0 H1 GLY 89
3 3.31 681 282 N GLY 37 N
4 3.56 1602 653 CT GLY 89 CA
5 3.67 152 0 H1 ASN 10
Connections and Nearest Atoms for NE HIS 35 NE2 ( P2A # 653 PDB# 274 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.33 651 273 CE HIS 35 CE1 1 HB 2.91 822 332 O MET 44 O
2 1.37 655 272 CD HIS 35 CD2 2 3.24 814 0 H1 MET 44
3 3.24 850 348 CD HIS 46 CD2
4 3.34 1593 0 H1 GLY 88
5 3.75 848 350 NE HIS 46 NE2
Suggestion: 2
3
Multiple AMBER options for HIS 46 ( 46) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 46 ND1 ( P2A # 844 PDB# 347 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.37 843 346 CG HIS 46 CG 1 2.62 2166 0 H1 MET 121
2 1.33 846 349 CE HIS 46 CE1 2 3.23 2080 863 ND HIS 117 ND1
3 2.04 2318 959 CU CU 130 CU 3 HB 3.33 2163 900 S MET 121 SD
4 3.40 2164 901 CT MET 121 CE
5 3.57 2082 865 CE HIS 117 CE1
Connections and Nearest Atoms for NE HIS 46 NE2 ( P2A # 848 PDB# 350 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.32 846 349 CE HIS 46 CE1 1 HB 2.70 162 67 O ASN 10 O
2 1.37 850 348 CD HIS 46 CD2 2 2.83 814 0 H1 MET 44
3 3.23 2166 0 H1 MET 121
4 3.52 822 332 O MET 44 O
5 3.74 813 334 CT MET 44 CG
Suggestion: 2
Enter
Multiple AMBER options for HIS 83 ( 83) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 83 ND1 ( P2A # 1494 PDB# 613 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.39 1493 612 CG HIS 83 CG 1 2.67 1317 0 HC VAL 73
2 1.33 1496 615 CE HIS 83 CE1 2 3.63 1315 542 CT VAL 73 CG2
3 3.74 1310 0 HC VAL 73
4 3.82 1316 0 HC VAL 73
5 3.86 1313 0 HC VAL 73
Connections and Nearest Atoms for NE HIS 83 NE2 ( P2A # 1498 PDB# 616 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.32 1496 615 CE HIS 83 CE1 1 3.09 1313 0 HC VAL 73
2 1.38 1500 614 CD HIS 83 CD2 2 3.44 1317 0 HC VAL 73
3 3.88 2385 981 O HOH 152 O
4 3.93 1311 541 CT VAL 73 CG1
5 4.03 1309 540 CT VAL 73 CB
Suggestion: 2
3
Multiple AMBER options for CYS 112 ( 112) :
1 CYS Cysteine (SH)
2 CYM Deprotonated Cysteine (S-)
3 CYX Cystine (S-S bridge)
Connections and Nearest Atoms for SG CYS 112 SG ( P2A # 2001 PDB# 828 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.82 1998 827 CB CYS 112 CB 1 2.53 858 0 H ASN 47
2 2.14 2318 959 CU CU 130 CU 2 2.65 2023 0 H PHE 114
3 3.00 2028 0 HC PHE 114
4 3.29 868 0 H ASN 47
5 3.39 2027 0 HC PHE 114
Suggestion: 2
Enter
Multiple AMBER options for HIS 117 ( 117) :
1 HID Histidine Delta Hydrogen
2 HIE Histidine Epsilon Hydrogen
3 HIP Histidine E & D Hydrogens
Connections and Nearest Atoms for ND HIS 117 ND1 ( P2A # 2080 PDB# 863 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.37 2079 862 CG HIS 117 CG 1 2.82 2028 0 HC PHE 114
2 1.34 2082 865 CE HIS 117 CE1 2 3.23 844 347 ND HIS 46 ND1
3 1.99 2318 959 CU CU 130 CU 3 3.26 2031 0 HA PHE 114
4 3.27 832 340 O GLY 45 O
5 3.43 846 349 CE HIS 46 CE1
Connections and Nearest Atoms for NE HIS 117 NE2 ( P2A # 2084 PDB# 866 )
Dist P2A Nr PDB Nr Label Near Dist P2A Nr PDB Nr Label
1 1.31 2082 865 CE HIS 117 CE1 1 2.57 209 0 H1 MET 13
2 1.37 2086 864 CD HIS 117 CD2 2 2.65 2031 0 HA PHE 114
3 HB 2.74 2406 988 O HOH 159 O
4 3.34 2030 841 CA PHE 114 CD1
5 3.41 204 0 H1 MET 13
Suggestion: 2
Enter
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Making Choices for Chain 1
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Completing residues with multiple options available, and solvent molecules
Checking positions of newly added atoms
Making choice for which molecules should be QM, which MM
Residues belonging to chain 0
Option Molecule Option Molecule Option Molecule Option Molecule Option Molecule
1: ALA 1 28: GLN 28 55: ASP 55 82: ALA 82 109: ALA 109
2: GLN 2 29: PHE 29 56: LYS 56 83: HIS 83 110: TYR 110
3: CYS 3 30: THR 30 57: GLN 57 84: THR 84 111: PHE 111
4: GLU 4 31: MET 31 58: ALA 58 85: LYS 85 112: CYS 112
5: ALA 5 32: HIS 32 59: VAL 59 86: VAL 86 113: SER 113
6: THR 6 33: LEU 33 60: ALA 60 87: ILE 87 114: PHE 114
7: VAL 7 34: LYS 34 61: THR 61 88: GLY 88 115: PRO 115
8: GLU 8 35: HIS 35 62: ASP 62 89: GLY 89 116: GLY 116
9: SER 9 36: VAL 36 63: GLY 63 90: GLY 90 117: HIS 117
10: ASN 10 37: GLY 37 64: MET 64 91: GLU 91 118: TRP 118
11: ASP 11 38: LYS 38 65: GLY 65 92: SER 92 119: ALA 119
12: ALA 12 39: MET 39 66: ALA 66 93: ASP 93 120: MET 120
13: MET 13 40: ALA 40 67: GLY 67 94: SER 94 121: MET 121
14: GLN 14 41: LYS 41 68: LEU 68 95: VAL 95 122: LYS 122
15: TYR 15 42: VAL 42 69: ALA 69 96: THR 96 123: GLY 123
16: ASN 16 43: ALA 43 70: GLN 70 97: PHE 97 124: THR 124
17: VAL 17 44: MET 44 71: ASP 71 98: ASP 98 125: LEU 125
18: LYS 18 45: GLY 45 72: TYR 72 99: VAL 99 126: LYS 126
19: GLU 19 46: HIS 46 73: VAL 73 100: SER 100 127: LEU 127
20: ILE 20 47: ASN 47 74: LYS 74 101: LYS 101 128: GLY 128
21: VAL 21 48: LEU 48 75: ALA 75 102: ILE 102 129: SER 129
22: VAL 22 49: VAL 49 76: GLY 76 103: ALA 103 130: CU 130
23: ASP 23 50: LEU 50 77: ASP 77 104: ALA 104
24: LYS 24 51: THR 51 78: THR 78 105: GLY 105
25: SER 25 52: LYS 52 79: ARG 79 106: GLU 106
26: CYS 26 53: ASP 53 80: VAL 80 107: ASN 107
27: LYS 27 54: ALA 54 81: ILE 81 108: TYR 108
Give option number of molecules to be put in QM region (or 'c' to continue):
Note: by specifying a negative number a molecule is removed from the QM region
45 46 112 117 121 130
Putting GLY 45 in QM region
Putting HIS 46 in QM region
Putting CYS 112 in QM region
Putting HIS 117 in QM region
Putting MET 121 in QM region
Putting CU 130 in QM region
Give option number of molecules to be put in QM region (or 'c' to continue):
Note: by specifying a negative number a molecule is removed from the QM region
c
Make a choice for the QM/MM treatment of GLY 45
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 2
Give choice:
Enter
Make a choice for the QM/MM treatment of HIS 46
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 1
Give choice:
Enter
Make a choice for the QM/MM treatment of CYS 112
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 3
Give choice:
Enter
Make a choice for the QM/MM treatment of HIS 117
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 3
Give choice:
Enter
Make a choice for the QM/MM treatment of MET 121
0: Put completely in QM region
1: Cut off at C-alpha (put NH in QM region, CO in MM region)
2: Cut off at C-alpha (put NH in MM region, CO in QM region)
3: Cut off at C-alpha (put NH and CO in MM region)
4: Cut off at C-alpha (put NH and CO in QM region, sidechain in MM region)
5: Put only part of sidechain in QM region
Suggestion: 3
Give choice:
Enter
Make a choice for the QM/MM treatment of CU 130
0: Put completely in QM region
1: Put only part of molecule in QM region
Suggestion: 0
Give choice:
Enter
Total formal charge on molecule CU 130 2.0000
Solvent molecules (SOL/HOH) belonging to this chain:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
61 62 63 64 65 66
Give the number of the molecule to be put in QM region (or 'c' to continue):
c
Residues belonging to chain 1
Do you want to add solvent to your system (Y/n) ?
Enter
Solvent (box) available:
1: HOH HOH Water molecule
2: MOH MOH Methanol molecule
3: CHL CHL Chloroform molecule
1
Reading contents of solvent box p2abox.HOH
Box Shape options:
1 Spherical box
2 Cubic box
Make a choice:
1
Writing inputfile for chain 0
Using total charge 1.0 and total spin 1.0
Maximum atomic distance (Angs) from center 25.62
Give boxsize (def.: 28.62 Angs)
30.0
Using BOXSIZE value of 30.0000
Adding atoms for box 1 Added (Box): 0 (Total): 0 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 2 Added (Box): 9 (Total): 9 Excl. (1): 639 Excl. (2): 0
Adding atoms for box 3 Added (Box): 3 (Total): 12 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 4 Added (Box): 0 (Total): 12 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 5 Added (Box): 6 (Total): 18 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 6 Added (Box): 228 (Total): 246 Excl. (1): 420 Excl. (2): 0
Adding atoms for box 7 Added (Box): 219 (Total): 465 Excl. (1): 429 Excl. (2): 0
Adding atoms for box 8 Added (Box): 9 (Total): 474 Excl. (1): 639 Excl. (2): 0
Adding atoms for box 9 Added (Box): 0 (Total): 474 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 10 Added (Box): 225 (Total): 699 Excl. (1): 423 Excl. (2): 0
Adding atoms for box 11 Added (Box): 216 (Total): 915 Excl. (1): 432 Excl. (2): 0
Adding atoms for box 12 Added (Box): 6 (Total): 921 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 13 Added (Box): 0 (Total): 921 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 14 Added (Box): 6 (Total): 927 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 15 Added (Box): 12 (Total): 939 Excl. (1): 636 Excl. (2): 0
Adding atoms for box 16 Added (Box): 0 (Total): 939 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 17 Added (Box): 12 (Total): 951 Excl. (1): 636 Excl. (2): 0
Adding atoms for box 18 Added (Box): 210 (Total): 1161 Excl. (1): 438 Excl. (2): 0
Adding atoms for box 19 Added (Box): 219 (Total): 1380 Excl. (1): 429 Excl. (2): 0
Adding atoms for box 20 Added (Box): 3 (Total): 1383 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 21 Added (Box): 216 (Total): 1599 Excl. (1): 417 Excl. (2): 15
Adding atoms for box 22 Added (Box): 381 (Total): 1980 Excl. (1): 3 Excl. (2): 264
Adding atoms for box 23 Added (Box): 261 (Total): 2241 Excl. (1): 3 Excl. (2): 384
Adding atoms for box 24 Added (Box): 183 (Total): 2424 Excl. (1): 423 Excl. (2): 42
Adding atoms for box 25 Added (Box): 189 (Total): 2613 Excl. (1): 426 Excl. (2): 33
Adding atoms for box 26 Added (Box): 186 (Total): 2799 Excl. (1): 3 Excl. (2): 459
Adding atoms for box 27 Added (Box): 351 (Total): 3150 Excl. (1): 3 Excl. (2): 294
Adding atoms for box 28 Added (Box): 222 (Total): 3372 Excl. (1): 420 Excl. (2): 6
Adding atoms for box 29 Added (Box): 9 (Total): 3381 Excl. (1): 639 Excl. (2): 0
Adding atoms for box 30 Added (Box): 162 (Total): 3543 Excl. (1): 429 Excl. (2): 57
Adding atoms for box 31 Added (Box): 219 (Total): 3762 Excl. (1): 426 Excl. (2): 3
Adding atoms for box 32 Added (Box): 6 (Total): 3768 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 33 Added (Box): 6 (Total): 3774 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 34 Added (Box): 219 (Total): 3993 Excl. (1): 426 Excl. (2): 3
Adding atoms for box 35 Added (Box): 216 (Total): 4209 Excl. (1): 432 Excl. (2): 0
Adding atoms for box 36 Added (Box): 6 (Total): 4215 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 37 Added (Box): 219 (Total): 4434 Excl. (1): 426 Excl. (2): 3
Adding atoms for box 38 Added (Box): 279 (Total): 4713 Excl. (1): 6 Excl. (2): 363
Adding atoms for box 39 Added (Box): 231 (Total): 4944 Excl. (1): 0 Excl. (2): 417
Adding atoms for box 40 Added (Box): 195 (Total): 5139 Excl. (1): 432 Excl. (2): 21
Adding atoms for box 41 Added (Box): 231 (Total): 5370 Excl. (1): 414 Excl. (2): 3
Adding atoms for box 42 Added (Box): 324 (Total): 5694 Excl. (1): 0 Excl. (2): 324
Adding atoms for box 43 Added (Box): 408 (Total): 6102 Excl. (1): 6 Excl. (2): 234
Adding atoms for box 44 Added (Box): 204 (Total): 6306 Excl. (1): 435 Excl. (2): 9
Adding atoms for box 45 Added (Box): 6 (Total): 6312 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 46 Added (Box): 177 (Total): 6489 Excl. (1): 435 Excl. (2): 36
Adding atoms for box 47 Added (Box): 219 (Total): 6708 Excl. (1): 429 Excl. (2): 0
Adding atoms for box 48 Added (Box): 6 (Total): 6714 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 49 Added (Box): 0 (Total): 6714 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 50 Added (Box): 3 (Total): 6717 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 51 Added (Box): 6 (Total): 6723 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 52 Added (Box): 0 (Total): 6723 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 53 Added (Box): 9 (Total): 6732 Excl. (1): 639 Excl. (2): 0
Adding atoms for box 54 Added (Box): 222 (Total): 6954 Excl. (1): 426 Excl. (2): 0
Adding atoms for box 55 Added (Box): 213 (Total): 7167 Excl. (1): 426 Excl. (2): 9
Adding atoms for box 56 Added (Box): 6 (Total): 7173 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 57 Added (Box): 3 (Total): 7176 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 58 Added (Box): 219 (Total): 7395 Excl. (1): 423 Excl. (2): 6
Adding atoms for box 59 Added (Box): 219 (Total): 7614 Excl. (1): 429 Excl. (2): 0
Adding atoms for box 60 Added (Box): 6 (Total): 7620 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 61 Added (Box): 0 (Total): 7620 Excl. (1): 648 Excl. (2): 0
Adding atoms for box 62 Added (Box): 12 (Total): 7632 Excl. (1): 636 Excl. (2): 0
Adding atoms for box 63 Added (Box): 3 (Total): 7635 Excl. (1): 645 Excl. (2): 0
Adding atoms for box 64 Added (Box): 0 (Total): 7635 Excl. (1): 648 Excl. (2): 0
Writing inputfile for chain 1
There are no atoms in this chain, ignoring it
Inputfile(s) written, everything processed, work has been done.
Thank you for using the PDB2ADF program.
================================
Normal ending of PDB2ADF program
================================
Contents of the 1DYZ.pdb2adf file generated by pdb2adf¶
The file is not given completely, since it contains more than 9000 atoms. Note that the NEWQMMM format is used if the environment variable SCM_PDB2ADF is set to NEW. This example uses the default ‘old’ QM/MM input style.
#! /bin/sh
$ADFBIN/adf << eor
TITLE QM/MM calculation setup by pdb2adf: M.Swart, 2005
Symmetry NOSYM
EPRINT
SFO NOEIG NOOVL
END
XC
GGA Becke-Perdew
END
GEOMETRY
CONVERGE grad=1.0e-3 rad=1.0e-1
END
BASIS
type TZP
core small
END
SCF
Converge 1.0e-5 1.0e-5
Iterations 99
END
INTEGRATION 5.0 5.0 5.0
CHARGE 1.0 1.0
UNRESTRICTED
ATOMS
1 C 11.3760 8.5410 29.7530
2 H 10.9114 9.3322 30.3413
3 H 12.4602 8.6423 29.8009
4 C 10.9630 8.7450 28.3090
5 O 10.8510 7.7910 27.5300
6 N 10.6890 9.9800 27.9260
7 H 10.7572 10.7382 28.5898
8 C 10.2900 10.2500 26.5530
9 H 10.5517 9.3991 25.9240
10 C 8.7770 10.5120 26.4440
11 H 8.5050 11.3473 27.0893
12 H 8.5229 10.7532 25.4118
13 C 7.9110 9.3590 26.8430
14 N 8.0710 8.0910 26.3490
15 C 7.1230 7.3010 26.8370
16 H 7.0894 6.2496 26.5516
17 N 6.3580 8.0230 27.6330
18 H 5.5568 7.6742 28.1395
19 C 6.8210 9.3110 27.6620
20 H 6.3141 10.0588 28.2719
21 C 11.0290 8.8020 20.9600
22 H 11.3902 9.8061 21.1823
23 C 10.0620 8.3640 22.0630
24 H 9.2477 9.0845 22.1402
25 H 9.6557 7.3817 21.8218
26 S 10.8340 8.2410 23.7100
27 C 10.1650 3.3080 22.4340
28 H 9.2929 2.7403 22.7584
29 C 10.1750 4.6030 23.2620
30 H 11.1220 5.1220 23.1143
31 H 9.3551 5.2459 22.9418
32 C 10.0160 4.3980 24.7440
33 N 9.7040 5.4090 25.6080
34 C 9.6570 4.9300 26.8540
35 H 9.4228 5.5952 27.6851
36 N 9.9280 3.6450 26.8000
37 H 9.9617 3.0260 27.5974
38 C 10.1580 3.2710 25.4990
39 H 10.3982 2.2340 25.2644
40 C 6.0350 6.2800 19.5280
41 H 4.9702 6.5113 19.5559
42 C 6.6730 6.7710 20.8330
43 H 7.7511 6.6157 20.7919
44 H 6.4641 7.8329 20.9631
45 C 6.1560 6.0500 22.0720
46 H 5.0693 6.1257 22.1101
47 H 6.4453 5.0000 22.0292
48 S 6.7760 6.6970 23.6140
49 C 6.0690 8.3070 23.6050
50 H 4.9825 8.2271 23.5709
51 H 6.3654 8.8396 24.5086
52 H 6.4202 8.8537 22.7299
53 CU 9.5640 7.3450 25.1750
54 N 10.9860 7.2480 30.2860
55 C 10.9790 11.4950 26.0450
56 N 10.3490 8.8040 19.6720
57 C 12.2010 7.8400 20.8870
58 N 11.3800 2.5190 22.6730
59 C 9.8250 3.6710 20.9970
60 N 6.2160 4.8460 19.4030
61 C 6.6330 7.0040 18.3530
62 N -1.1930 25.6890 17.1840
63 H -0.3133 25.1929 17.1970
64 H -1.3738 25.1438 18.0148
65 H -1.5170 24.8559 16.7138
66 C -1.4820 27.1340 16.8960
67 H -2.1350 27.2082 16.0264
68 C -2.1950 27.7860 18.0880
69 H -1.5602 27.7210 18.9717
70 H -2.3971 28.8331 17.8627
71 H -3.1350 27.2677 18.2776
72 C -0.1820 27.8790 16.5880
73 O 0.8890 27.4920 17.0690
74 N -0.2890 28.9420 15.7940
75 H -1.1936 29.2105 15.4339
76 C 0.8750 29.7430 15.4220
77 H 0.5616 30.5606 14.7728
78 C 1.5270 30.3290 16.6860
...
9746 O 31.1328 34.4612 22.6903
9747 H 31.8908 34.5740 22.1167
9748 H 30.6706 35.2981 22.6446
END
QMMM
FORCE_FIELD_FILE $ADFRESOURCES/ForceFields/amber95.ff
RESTART_FILE mm.restart ! old style restart
OUTPUT_LEVEL=1
WARNING_LEVEL=0
! -------------------
! for AddRemove model
! -------------------
PARTITION=5
ELSTAT_COUPLING_MODEL=4 ! for AddRemove model
OPTIMIZE
METHOD CONJGRAD
MAX_STEPS 5000
PRINT_CYCLES 10
MM_NOTCONVERGED 0
SUBEND
LINK_BONDS
1 - 54 1.3320 H H1
8 - 55 1.3861 H H1
21 - 56 1.3362 H H1
21 - 57 1.3927 H H1
27 - 58 1.3471 H H1
27 - 59 1.3951 H H1
40 - 60 1.3310 H H1
40 - 61 1.3799 H H1
SUBEND
MM_CONNECTION_TABLE
1 CT QM 54 4 2 3
2 H1 QM 1
3 H1 QM 1
4 C QM 1 5 6
5 O QM 4
6 N QM 8 4 7
7 H QM 6
8 CT QM 6 10 55 9
9 H1 QM 8
10 CT QM 8 13 11 12
11 HC QM 10
12 HC QM 10
13 CC QM 10 14 19
14 NB QM 13 15 53
15 CR QM 14 17 16
16 H5 QM 15
17 NA QM 15 19 18
18 H QM 17
19 CW QM 13 17 20
20 H4 QM 19
21 CT QM 56 23 57 22
22 H1 QM 21
23 CT QM 21 26 24 25
24 H1 QM 23
25 H1 QM 23
26 SH QM 23 53
27 CT QM 58 29 59 28
28 H1 QM 27
29 CT QM 27 32 30 31
30 HC QM 29
31 HC QM 29
32 CC QM 29 33 38
33 NB QM 32 34 53
34 CR QM 33 36 35
35 H5 QM 34
36 NA QM 34 38 37
37 H QM 36
38 CW QM 32 36 39
39 H4 QM 38
40 CT QM 60 42 61 41
41 H1 QM 40
42 CT QM 40 45 43 44
43 HC QM 42
44 HC QM 42
45 CT QM 42 48 46 47
46 H1 QM 45
47 H1 QM 45
48 S QM 45 49
49 CT QM 48 50 51 52
50 H1 QM 49
51 H1 QM 49
52 H1 QM 49
53 CU QM 14 26 33
54 N LI 1 748 750
55 C LI 8 751 752
56 N LI 21 1683 1685
57 C LI 21 1686 1687
58 N LI 27 1737 1739
59 C LI 27 1740 1741
60 N LI 40 1790 1792
61 C LI 40 1793 1794
62 N3 MM 66 63 64 65
63 H MM 62
64 H MM 62
65 H MM 62
66 CT MM 62 68 72 67
67 HP MM 66
68 CT MM 66 69 70 71
69 HC MM 68
70 HC MM 68
71 HC MM 68
72 C MM 66 73 74
73 O MM 72
74 N MM 76 72 75
75 H MM 74
76 CT MM 74 78 89 77
77 H1 MM 76
78 CT MM 76 81 79 80
....
9746 OW MM 9747 9748
9747 HW MM 9746
9748 HW MM 9746
SUBEND
CHARGES
1 -0.0252
2 0.0698
3 0.0698
4 0.5973
5 -0.5679
6 -0.4157
7 0.2719
8 -0.0581
9 0.1360
10 -0.0074
11 0.0367
12 0.0367
13 0.1868
14 -0.5432
15 0.1635
16 0.1435
17 -0.2795
18 0.3339
19 -0.2207
20 0.1862
21 0.0350
22 0.0480
23 -0.7360
24 0.2440
25 0.2440
26 -0.7360
27 -0.0581
28 0.1360
29 -0.0074
30 0.0367
31 0.0367
32 0.1868
33 -0.5432
34 0.1635
35 0.1435
36 -0.2795
37 0.3339
38 -0.2207
39 0.1862
40 -0.0237
41 0.0880
42 0.0342
43 0.0241
44 0.0241
45 0.0018
46 0.0440
47 0.0440
48 -0.2737
49 -0.0536
50 0.0684
51 0.0684
52 0.0684
53 2.0000
54 -0.4157
55 0.5973
56 -0.4630
57 0.6160
58 -0.4157
59 0.5973
60 -0.4157
61 0.5973
62 0.1414
63 0.1997
64 0.1997
65 0.1997
66 0.0962
67 0.0889
68 -0.0597
69 0.0300
70 0.0300
71 0.0300
72 0.6163
73 -0.5722
74 -0.4157
75 0.2719
76 -0.0031
77 0.0850
78 -0.0036
....
9746 -0.8340
9747 0.4170
9748 0.4170
SUBEND
END
COMMENT
Atom ADF ID p2a ID Amber Mol Nr Amber pdb ID atom
C 1 828 CT GLY 45 GLY 338 CA
H 2 829 H1 GLY 45 GLY 0 HA2
H 3 830 H1 GLY 45 GLY 0 HA3
C 4 831 C GLY 45 GLY 339 C
O 5 832 O GLY 45 GLY 340 O
N 6 836 N HIS 46 HIE 341 N
H 7 837 H HIS 46 HIE 0 H
C 8 838 CT HIS 46 HIE 342 CA
H 9 839 H1 HIS 46 HIE 0 HA
C 10 840 CT HIS 46 HIE 345 CB
H 11 841 HC HIS 46 HIE 0 HB2
H 12 842 HC HIS 46 HIE 0 HB3
C 13 843 CC HIS 46 HIE 346 CG
N 14 844 NB HIS 46 HIE 347 ND1
C 15 846 CR HIS 46 HIE 349 CE1
C 15 846 CR HIS 46 HIE 349 CE1
H 16 847 H5 HIS 46 HIE 0 HE1
N 17 848 NA HIS 46 HIE 350 NE2
H 18 849 H HIS 46 HIE 0 HE2
C 19 850 CW HIS 46 HIE 348 CD2
H 20 851 H4 HIS 46 HIE 0 HD2
C 21 1996 CT CYS 112 CYM 824 CA
H 22 1997 H1 CYS 112 CYM 0 HA
C 23 1998 CT CYS 112 CYM 827 CB
H 24 1999 H1 CYS 112 CYM 0 HB3
H 25 2000 H1 CYS 112 CYM 0 HB2
S 26 2001 SH CYS 112 CYM 828 SG
C 27 2074 CT HIS 117 HIE 858 CA
H 28 2075 H1 HIS 117 HIE 0 HA
C 29 2076 CT HIS 117 HIE 861 CB
H 30 2077 HC HIS 117 HIE 0 HB2
H 31 2078 HC HIS 117 HIE 0 HB3
C 32 2079 CC HIS 117 HIE 862 CG
N 33 2080 NB HIS 117 HIE 863 ND1
C 34 2082 CR HIS 117 HIE 865 CE1
H 35 2083 H5 HIS 117 HIE 0 HE1
N 36 2084 NA HIS 117 HIE 866 NE2
H 37 2085 H HIS 117 HIE 0 HE2
C 38 2086 CW HIS 117 HIE 864 CD2
H 39 2087 H4 HIS 117 HIE 0 HD2
C 40 2155 CT MET 121 MET 895 CA
H 41 2156 H1 MET 121 MET 0 HA
C 42 2157 CT MET 121 MET 898 CB
H 43 2158 HC MET 121 MET 0 HB2
H 44 2159 HC MET 121 MET 0 HB3
C 45 2160 CT MET 121 MET 899 CG
H 46 2161 H1 MET 121 MET 0 HG2
H 47 2162 H1 MET 121 MET 0 HG3
S 48 2163 S MET 121 MET 900 SD
C 49 2164 CT MET 121 MET 901 CE
H 50 2165 H1 MET 121 MET 0 HE1
H 51 2166 H1 MET 121 MET 0 HE2
H 52 2167 H1 MET 121 MET 0 HE3
CU 53 2318 CU CU 130 959 CU
N 54 826 N GLY 45 GLY 337 N
C 55 852 C HIS 46 HIE 343 C
N 56 1994 N CYS 112 CYM 823 N
C 57 2003 C CYS 112 CYM 825 C
N 58 2072 N HIS 117 HIE 857 N
C 59 2088 C HIS 117 HIE 859 C
N 60 2153 N MET 121 MET 894 N
C 61 2168 C MET 121 MET 896 C
N 62 1 N3 ALA 1 ALA 1 N
H 63 2 H ALA 1 ALA 0 H1
H 64 11 H ALA 1 ALA 0 H2
H 65 12 H ALA 1 ALA 0 H3
C 66 3 CT ALA 1 ALA 2 CA
H 67 4 HP ALA 1 ALA 0 HA
C 68 5 CT ALA 1 ALA 5 CB
H 69 6 HC ALA 1 ALA 0 HB1
H 70 7 HC ALA 1 ALA 0 HB2
H 71 8 HC ALA 1 ALA 0 HB3
C 72 9 C ALA 1 ALA 3 C
O 73 10 O ALA 1 ALA 4 O
N 74 14 N GLN 2 GLN 6 N
H 75 15 H GLN 2 GLN 0 H
C 76 16 CT GLN 2 GLN 7 CA
H 77 17 H1 GLN 2 GLN 0 HA
C 78 18 CT GLN 2 GLN 10 CB
....
O 2111 2517 OW HOH 196 SOL 1025 O
H 2112 2518 HW HOH 196 SOL 0 H1
H 2113 2519 HW HOH 196 SOL 0 H2
END
ENDINPUT
eor
An example on solvent shell run¶
The idea of this example is to make an adf-input file using a PDB file of water (hoh.pdb), in the solvent methanol. The water molecule in the adf-input file should be in the QM part, and the solvent methanol (in a box) is in MM part.
Contents of the hoh.pdb file¶
TITLE PDB-FILE CORRESPONDING TO pdb2adf-GENERATED ADF-INPUTFILE
REMARK Written by M. Swart, March 2005
HETATM 1 H1 HOH 1 1.716 26.282 11.239 1.00 0.00 1DYZ H
HETATM 2 O HOH 1 2.439 25.795 11.634 1.00 0.00 1DYZ O
HETATM 3 H2 HOH 1 3.140 26.440 11.729 1.00 0.00 1DYZ H
END
Usage of pdb2adf¶
The program works interactively. Given below in bold are the parts that the user has to type. In cases where the user agrees with the suggestion given by the program, the user can press the Enter key indicated with Enter.
P D B 2 A D F - program
version 2005.01
Written by: Marcel Swart, 2005
This program uses AMBER parameter files
see: http://amber.scripps.edu
Do you want a logfile to be written (Y/n) ?
Enter
Please give name of PDB-file
hoh.pdb
read fragments
Data Processed:
Nat: 3
Nmol: 1
NChains: 0
Please wait, making connection tables
Now finding nearby atoms
Assigning chain ID to all residues
Completing residues for which only option is available
Refinding nearby atoms (including atoms added in residue completion)
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Making Choices for Chain 0
- - - - - - - - - - - - - - - - - - - - - - - - - - -
Completing residues with multiple options available, and solvent molecules
Checking positions of newly added atoms
Making choice for which molecules should be QM, which MM
Residues belonging to chain 0
Solvent molecules (SOL/HOH) belonging to this chain:
1
Give the number of the molecule to be put in QM region (or 'c' to continue):
1
Putting HOH 1 in QM region
Give the number of the molecule to be put in QM region (or 'c' to continue):
c
Do you want to add solvent to your system (Y/n) ?
Enter
Solvent (box) available:
1: HOH HOH Water molecule
2: MOH MOH Methanol molecule
3: CHL CHL Chloroform molecule
2
Reading contents of solvent box p2abox.MOH
Box Shape options:
1 Spherical box
2 Cubic box
Make a choice:
1
Writing inputfile for chain 0
Using total charge 0.0 and total spin 0.0
Maximum atomic distance (Angs) from center 0.92
Give boxsize (def.: 15.00 Angs)
14.0
Using BOXSIZE value of 14.0000
Adding atoms for box 1 Added (Box): 84 (Total): 84 Excl. (1): 660 Excl. (2): 6
Adding atoms for box 2 Added (Box): 102 (Total): 186 Excl. (1): 642 Excl. (2): 6
Adding atoms for box 3 Added (Box): 102 (Total): 288 Excl. (1): 642 Excl. (2): 6
Adding atoms for box 4 Added (Box): 108 (Total): 396 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 5 Added (Box): 120 (Total): 516 Excl. (1): 630 Excl. (2): 0
Adding atoms for box 6 Added (Box): 96 (Total): 612 Excl. (1): 654 Excl. (2): 0
Adding atoms for box 7 Added (Box): 108 (Total): 720 Excl. (1): 642 Excl. (2): 0
Adding atoms for box 8 Added (Box): 102 (Total): 822 Excl. (1): 642 Excl. (2): 6
Inputfile(s) written, everything processed, work has been done.
Thank you for using the PDB2ADF program.
================================
Normal ending of PDB2ADF program
================================
Contents of the hoh.pdb2adf file generated by pdb2adf¶
The file is not given completely, since it contains more than 800 atoms. Note that the NEWQMMM format is used if the environment variable SCM_PDB2ADF is set to NEW. This example uses the default ‘old’ QM/MM input style.
#! /bin/sh
$ADFBIN/adf << eor
TITLE QM/MM calculation setup by pdb2adf: M.Swart, 2005
Symmetry NOSYM
EPRINT
SFO NOEIG NOOVL
END
XC
GGA Becke-Perdew
END
GEOMETRY
CONVERGE grad=1.0e-3 rad=1.0e-1
END
BASIS
type TZP
core small
END
SCF
Converge 1.0e-5 1.0e-5
Iterations 99
END
INTEGRATION 5.0 5.0 5.0
CHARGE 0.0
ATOMS
1 O 2.4390 25.7950 11.6340
2 H 1.7160 26.2820 11.2390
3 H 3.1400 26.4400 11.7290
4 C -10.0667 22.2493 11.7437
5 H -10.2077 21.5053 10.9597
6 H -10.5047 21.8683 12.6667
7 H -10.5167 23.2103 11.4977
8 O -8.7387 22.3983 12.0617
9 H -8.3007 22.6943 11.2607
10 C -0.2827 19.0253 2.2847
11 H -0.5357 18.2063 2.9567
12 H 0.7633 19.2913 2.4407
13 H -0.9267 19.8753 2.5107
14 O -0.4997 18.6373 0.9467
15 H 0.1123 17.9313 0.7287
....
823 H 5.4711 27.9401 19.5645
824 O 5.5611 28.7181 17.7095
825 H 5.2631 27.8621 17.3935
END
QMMM
FORCE_FIELD_FILE $ADFRESOURCES/ForceFields/amber95.ff
RESTART_FILE mm.restart ! old style restart
OUTPUT_LEVEL=1
WARNING_LEVEL=0
! -------------------
! for AddRemove model
! -------------------
PARTITION=5
ELSTAT_COUPLING_MODEL=4 ! for AddRemove model
OPTIMIZE
METHOD CONJGRAD
MAX_STEPS 5000
PRINT_CYCLES 10
MM_NOTCONVERGED 0
SUBEND
MM_CONNECTION_TABLE
1 OW QM 2 3
2 HW QM 1
3 HW QM 1
4 CT MM 5 6 7 8
5 H1 MM 4
6 H1 MM 4
7 H1 MM 4
8 OH MM 4 9
9 HO MM 8
10 CT MM 11 12 13 14
11 H1 MM 10
12 H1 MM 10
13 H1 MM 10
14 OH MM 10 15
15 HO MM 14
....
823 H1 MM 820
824 OH MM 820 825
825 HO MM 824
SUBEND
CHARGES
1 -0.8340
2 0.4170
3 0.4170
4 0.1166
5 0.0372
6 0.0372
7 0.0372
8 -0.6497
9 0.4215
10 0.1166
11 0.0372
12 0.0372
13 0.0372
14 -0.6497
15 0.4215
...
823 0.0372
824 -0.6497
825 0.4215
SUBEND
END
COMMENT
Atom ADF ID p2a ID Amber Mol Nr Amber pdb ID atom
O 1 1 OW HOH 1 SOL 2 O
H 2 2 HW HOH 1 SOL 1 H1
H 3 3 HW HOH 1 SOL 3 H2
END
ENDINPUT
eor