Technical topics¶
Input syntax¶
The AMS driver reads its input from standard input, i.e. what is called
STDIN on Unix-like systems. Technically it is possible to run AMS and type
the input file in interactively. This is however highly impractical and most
people run AMS from a small shell script that contains the AMS text input and
sends it directly to the AMS executable:
#!/bin/sh
$ADFBIN/ams << EOF
... AMS text input goes here:
Block
Keywork value
OtherKeyword value
End
EOF
This section of the AMS manual documents the syntax of the text input.
General remarks on input structure and parsing¶
Most keys are optionals. Defaults values will be used for keys that are not specified in the input
Keys/blocks can either be unique (i.e. they can appear in the input only once) or non-unique. (i.e. they can appear multiple times in the input)
The order in which keys or blocks are specified in the input does not matter. Possible exceptions to this rule are a) the content of non-standard blocks b) some non-unique keys/blocks)
Comments in the input file start with one of the following characters:
#,!,:::# this is a comment ! this is also a comment :: yet another comment
Empty lines are ignored
The input parsing is case insensitive (except for string values):
# this: UseSymmetry false # is equivalent to this: USESYMMETRY FALSE
Indentation does not matter and multiple spaces are treaded as a single space (except for string values):
# this: UseSymmetry false # is equivalent to this: UseSymmetry false
Keys¶
Key-value pairs have the following structure:
KeyName Value
Possible types of keys:
- bool key
The value is a single Boolean (logical) value. The value can be
True(equivalentlyYes) orFalse(equivalentlyNo.). Not specifying any value is equivalent to specifyingTrue. Example:KeyName Yes
- integer key
The value is a single integer number. Example:
KeyName 3
- float key
The value is a single float number. For scientific notation, the E-notation is used (e.g. \(-2.5 \times 10^{-3}\) can be expressed as
-2.5E-3). The decimal separator should be a dot (.), and not a comma (,). Example:KeyName -2.5E-3
- string key
The value is a string, which can include white spaces. Only ASCII characters are allowed. Example:
KeyName Lorem ipsum dolor sit amet
- multiple_choice key
The value should be a single word among the list options for that key (the options are listed in the documentation of the key). Example:
KeyName SomeOption
- integer_list key
The value is list of integer numbers. Example:
KeyName 1 6 0 9 -10
- float_list key
The value is list of float numbers. The convention for float numbers is the same as for Float keys. Example:
KeywordName 0.1 1.0E-2 1.3
Blocks¶
Blocks give a hierarchical structure to the input, grouping together related keys (and possibly sub-blocks). In the input, blocks generally span multiple lines, and have the following structure:
BlockName
KeyName1 value1
KeyName2 value2
...
End
Headers
For some blocks it is possible (or necessary) to specify a header next to the block name:
BlockName someHeader
KeyName1 value1
KeyName2 value2
...
End
Compact notation
It is possible to specify multiple key-value pairs of a block on a single line using the following notation:
# This:
BlockName KeyName1=value1 KeyName2=value2
# is equivalent to this:
BlockName
KeyName1 value1
KeyName2 value2
End
Notes on compact notation:
Spaces (blanks) between the key, the equal sign and the value are not allowed:
# This is OK: BlockName KeyName1=value1 KeyName2=value2 # This is NOT OK: BlockName KeyName1 = value1 KeyName2= value2
The compact notation can be used only for following types of keys: bool, integer, float and multiple_choice. It should not be used for sting, integer_list and float_list keys
The compact notation cannot be used for blocks with headers
input units cannot be defined in compact notation
Non-standard Blocks
A special type of block is the non-standard block. These blocks are used for parts of the input that do not follow the usual key-value paradigm.
A notable example of a non-standard block is the Atoms block (in which the atomic coordinates and atom types are defined).
Units¶
Some keys have a default unit associated (not all keys have units). For such keys, the default unit is mention in the key documentation. One can specify a different unit within square brackets at the end of the line:
KeyName value [unit]
For example, assuming the key EnergyThreshold has as default unit Hartree, then the following definitions are equivalent:
# Use defaults unit:
EnergyThreshold 1.0
# use eV as unit:
EnergyThreshold 27.211 [eV]
# use kcal/mol as unit:
EnergyThreshold 627.5 [kcal/mol]
# Hartree is the atomic unit of energy:
EnergyThreshold 1.0 [a.u.]
Available units:
- Energy:
Hartree(a.u.),eV,kJ/mol,kcal/mol,cm^-1 - Length:
Bohr(a.u.),Angstrom(A),nm,pm
Double parallelism¶
AMS is a parallel program using MPI for efficient execution on distributed memory machines, aka compute clusters. For most jobs, the AMS driver part of a calculation is computationally not particularly costly and most of the execution time is spent inside of the compute engines. Therefore the main parallelization of AMS is inside of the engines, making sure that a good performance is obtained for tasks such as molecular dynamics or geometry optimizations, which consist of a series of interdependent engine invocations: We need to have completed step \(n\) before we can continue with step \(n+1\).
However, not all workloads are of this sequentially dependent type. Some jobs have a lot of independent work, that can be done in parallel. This kind of trivial parallelizability can be exploited at the AMS driver level: Instead of having all cores collaborate on a single PES point and then doing all needed PES points sequentially, we can just distribute the available PES points over the all the available cores. Normally this leads to a better parallel scaling than the default parallelization inside of the engines: Parallelizing the engines is relatively complicated and often requires a lot of communication between cores. Parallelizing on the driver level on the other hand is very easy, and often the only communication required is at the very end of the calculation, when results are collected.
Note that it is perfectly possible to combine both the in-engine parallelization and the driver level parallelism: At the driver level we could split our e.g. in total 32 cores into 4 groups of 8 cores, and then have each group of 8 use the in-engine parallelization to collaborate on a specific calculation. This is especially useful if the total number of cores is larger than then number of independent calculations we have to do. It might also be that we have a very large number of calculations to do, but not enough memory to let every core work alone on its own calculation, as would be ideal from a parallel scaling point of view.
Because of the two levels of parallelism – both at the driver and the engine level – we call this setup double parallelization. In the 2018 release of AMS, double parallelization is used for the calculation of the PES point properties which are derivatives, if these need to be done numerically:
- Numerical calculation of forces / nuclear gradients. With a double sided derivative this requires \(6 \times n_\text{atoms}\) independent calculations on geometries with one atom displaced along a cartesian coordinate.
- Numerical calculation of the stress tensor for periodic systems. This requires up to 12 calculations for a double sided derivative along the 6 strain directions, but might require less in case some of the strains are symmetry equivalent.
- Numerical calculation of the Hessian and normal modes of vibration. This is currently only supported for engines that calculate nuclear gradients analytically and done by numerically differentiating this first (analytic) derivative. As such it requires \(6 \times n_\text{atoms}\) independent calculations on geometries with one atom displaced along a cartesian coordinate.
- Numerical calculation of the elastic tensor. This requires 84 independent geometry optimizations on systems with differently strained lattices, with each optimization having a variable number of steps.
- Numerical calculation of phonons. This requires at most \(6 \times n_\text{atoms}\) displacements, but might require less in case some of the displacements are symmetry equivalent. Note that the displacements are done in a super cell system, which for many engines will increase the memory requirements, but also improve the in-engine parallel scalability.
In order to use double parallelization it has to be enabled explicitly in the
input. This is done for the above mentioned properties individually, as one
might want a different grouping strategy for each property. For each property
there is a separate Parallel block somewhere in the input (e.g.
ElasticTensor%Parallel for the calculation of the elastic tensor), which
has the following keywords:
Parallel
nGroups integer
nCoresPerGroup integer
nNodesPerGroup integer
End
Note that only one of them should be specified in the input, depending of course on what is the desired strategy for parallelization.
nGroups n- Splits all cores evenly into
ngroups. We recommend choosingnsuch that it divides the total number of cores without a remainder. nCoresPerGroup n- Each group consists of
ncores. As suchnCoresPerGroup 1results in the maximum possible parallelism at the driver level. We recommend choosingnsuch that it divides the total number of cores without a remainder. nNodesPerGroup n- Makes groups from all cores within
nnodes, e.g.nNodesPerGroup 1would make every cluster node into a separate group. Note that this option should only be used on homogeneous compute clusters, where all used nodes have the same number of cores. Otherwise cores from different nodes will be grouped together in very surprising and unintended ways, probably resulting in suboptimal performance.
The optimal grouping strategy and number of groups depends on the total number of cores used in the calculation, the amount of independent tasks to be done in parallel, as well as the parallel scalability of the engine itself. In practice it can be a bit tricky. Suppose, as an example, that we want to calculate the elastic properties of a bulk material on a 32 core machine. The calculation of the elastic tensor should be done on a relaxed geometry, including relaxed lattice degrees of freedom. We therefore first perform a geometry optimization, before calculating the elastic tensor. In AMS this can easily be done with the following input:
Task GeometryOptimization
GeometryOptimization
OptimizeLattice True
End
Properties
ElasticTensor True
End
But what is the most optimal parallel setup for this calculation? First we recognize that performing a lattice optimization requires the calculation of the stress tensor at every step of the optimization. Assuming that our bulk system does not have any symmetries AMS can exploit, the numerical calculation of the stress tensor (which most engines can not calculate analytically) would require 12 independent strained calculations for every step in the geometry optimization. Once the geometry optimization is converged, we have to perform 84 independent geometry optimizations to determine the elements of the elastic tensor. In summary, the graph of dependencies between all these tasks looks like this:
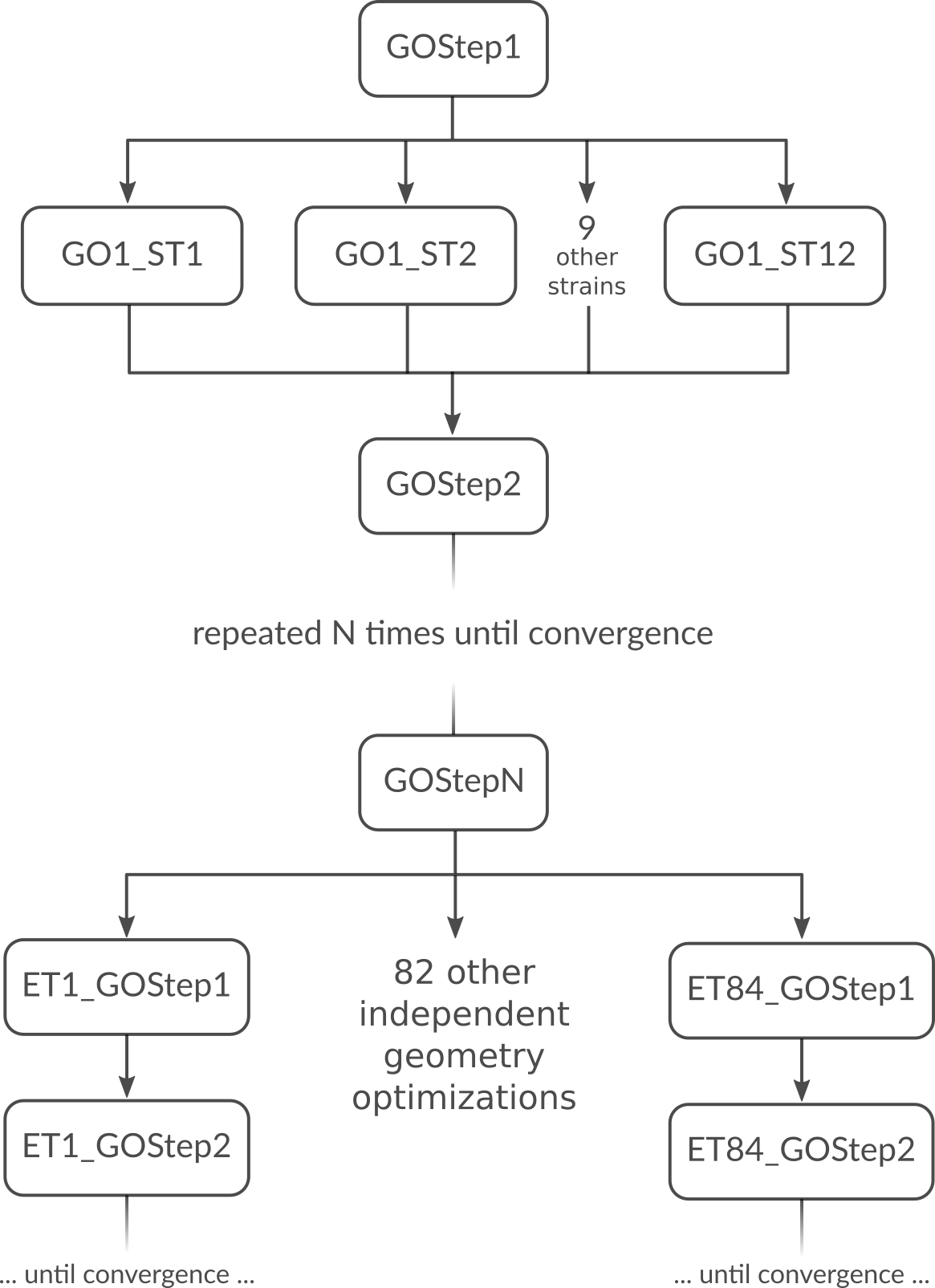
How do we best parallelize this? For the main steps, e.g. GOStep1 there is
no question: We have nothing to do in parallel and all 32 cores work on it
together to finish it as quickly as possible. For the numerical calculation of
the stress tensor we have 12 tasks that can be done in parallel by the 32 cores
in our machine. Now 12 obviously does not divide 32 without a remainder, so
there is no way to split into equally sized groups and do all 12 strains in
parallel. The greatest common divisor of 12 and 32 is 4, so it’s probably best
to split into 4 groups of 8 cores each. This is done with nGroups 4. Each
group would then do 3 of the 12 strained calculations sequentially, using the
in-engine parallelization to speed up the individual calculations. Once the
stress tensor is computed in this way all groups merge and all 32 cores work
together on GOStep2. This splitting and merging now continues until the
geometry optimization is converged. For the elastic tensor we now have 84 tasks
to perform in parallel, where each task is a completely separate geometry
optimization (without optimizing the lattice) of a strained system. 84 tasks is
more than double the number of cores we have. In this case it is probably best
to just run as parallel as possible at the driver level and make 32 “groups” of
just one core to throw the 84 tasks at. This is easily done by setting
nCoresPerGroup 1 in the ElasticTensor block. Putting everything together
we should add the following to our input file in order to optimally utilize our
machine for this example calculation:
NumericalDifferentiation
Parallel
nGroups 4
End
End
ElasticTensor
Parallel
nCoresPerGroup 1
End
End
Running AMS on compute clusters¶
AMS is parallelized with MPI and can therefore be run in parallel on distributed memory machines, aka compute clusters. See the installation manual for general documentation on how to set up and run all the programs from the Amsterdam Modeling Suite on compute clusters. In this section we give some more advice that is specific to the AMS driver and its engines.
Normally users use the login node to prepare their jobs and input files
somewhere in their home directory, and also want the results of their jobs to
end up there. Quite often, compute clusters are set up such that the user’s home
directory is also mounted on the compute nodes, usually via NFS (Network File
System). Before the introduction of the AMS driver it was not recommended to
cd to the home directory in the submission script and have the compute nodes
execute the job directly there. This was simply due to the fact that a lot of
file I/O was done on temporary files in the present working directory, which in
this case would be on a slow network-mounted file system.
On the other hand, with AMS, switching to the home directory is the preferred way of running on a cluster where the home directory is mounted on the compute nodes. Running in the home directory mounted over NFS does not come with a performance penalty for AMS, but has many advantages. This is because AMS and its engines are already built under the assumption that access to this directory is slow. Basically there are three directories that are used by the AMS driver and its engines:
- The starting directory, i.e. the present working directory at the time the AMS driver is started. This folder is generally read-only for AMS, except for creating the results directory there at the beginning of a calculation. Note that all relative paths in the AMS input, e.g. for loading results from previous calculations, are relative to the starting directory. The starting directory is assumed to be on a slow filesystem, but since data is normally only read once from there in the beginning of a calculation, this is in practice not a problem.
- The results directory, where the results of a calculation as well as important intermediate steps (e.g. restart files) are collected. It also contains the log file which can be used to monitor a running calculations. The results directory is assumed to be on a slow filesystem, so AMS and its engines will be very careful not to do much disk I/O there. Generally something is only written to the results directory when AMS is sure that it should remain on disk when the calculation finishes. The results directory can also contain some intermediate restart files, so the contents of the result directory should be all that is needed in case the calculation crashes or is killed before it finishes normally.
- The scratch directory, the location of which is set with the
$SCM_TMPDIRenvironment variable, see also the installation manual. This directory should be put on a fast disk, e.g. an SSD in the compute node, as it will be used to store temporary results on disk. Users do not really need to care or know about the temporary files in the scratch directory. Normally, any files and directories created in the scratch directory are cleaned up at the end of the calculation. In case of errors, AMS tries to copy anything useful (e.g. the text output of all the different ranks) to the results directory in order to make finding the problem easier. However, for some kinds of crashes (or if theSIGKILLsignal is sent to AMS), the cleanup of the scratch directory might not be performed, in which case users might want to manually check or remove theamstmp_*folders in the scratch directory.
With this setup there is no performance penalty for running directly on a network mounted home directory: Results will just be put there immediately, instead of being copied there at the end of a calculation.
Normally all batch systems provide an environment variable that is set to the
directory from which the job was submitted, which is then where one should
cd in the run script:
#!/bin/sh
if [ -z "$PBS_O_WORKDIR" ]; then
# PBS batch system
cd "$PBS_O_WORKDIR"
elif [ -z "$SLURM_SUBMIT_DIR" ]; then
# Slurm batch system
cd "$SLURM_SUBMIT_DIR"
elif [ -z "..." ]; then
# add other batch systems as necessary ...
cd "..."
fi
export AMS_JOBNAME=myJob
$ADFBIN/ams << EOF
# Normal AMS text input, but with all paths
# relative to where the job was submitted from, e.g.:
LoadSystem previousJob.results/ams.rkf
EOF
With this runscript the AMS driver would make a myJob.results folder in the
directory where the job was submitted from, and there is no need to copy results
around manually in the run script. Furthermore this runscript always produces
exactly the same files in the same locations, no matter if it is run
interactively or submitted to a compute node through the batch system.
Furthermore all paths in the input file can be specified relative to the
location from where the runscript is submitted (normally the folder in which the
runscript is located). This removes the need to copy or specify absolute paths
to previous results, e.g. when restarting calculations. Finally, files useful
for monitoring the running calculation are also conveniently there and not
hidden somewhere on the compute node.
Python interface¶
There is a complete Python interface to AMS, which allows users to set up and run arbitrary AMS jobs, and to conveniently analyze the calculation results directly from Python. In this way AMS jobs can be automatized and complex multi-stage workflows implemented.
The scripting framework is called PLAMS as in “Python Library for Automating Molecular Simulation”, which conveniently can also be read as “Python Layer for AMS”. It is documented in a separate manual: