Example: The role of specifying the atom types¶
Now we look at a Propanenitrile molecule, the QM region is highlighted.
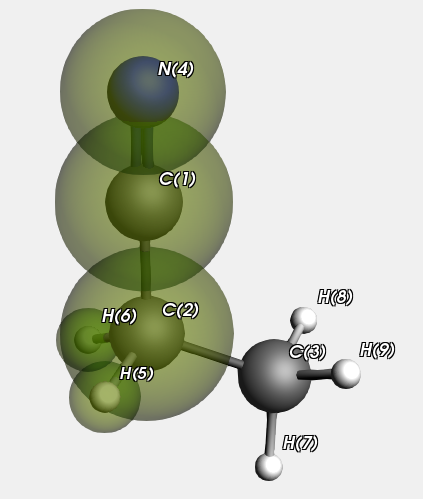
We run this with and without specifying the atom types on input. In principle this makes a difference for the MM type for atom “C(3)” in the MM sub calculation on atoms 3,7,8, and 9. If specified it will be C_3 (as it is in the whole Propanenitrile molecule), but if not it will be guessed as C_R. In practice there is no effect for this calculation.
Let us have a look at the report generated by the example, that pretty much explains what is done
Download report Propanenitrile.txt
We first check how bad the MM method is compared to the QM method for some distances in the QM region.
Here are the distances (Angstrom) as obtained with a QM and an MM method
Distance QM MM Err(MM)
-------------------------------------------
C(1)-C(2) 1.456 1.467 0.011
C(1)-N(4) 1.147 1.157 0.010
C(2)-H(5) 1.095 1.110 0.015
Can we get better results for the QM region with the hybrid engine?
Even though UFF has automatic atom typing, it still matters (in principle) whether we specify it on input or not:
* Without typing for each region the types are automatically guessed
* With typing the types are always as on input (for all regions)
The only difference is in the C type for the MM region.
Here are the distances (Angstrom) as obtained with a QM and an Hybrid method without explicit typing:
Distance QM MM Err(MM)
-------------------------------------------
C(1)-C(2) 1.456 1.456 0.000
C(1)-N(4) 1.147 1.147 -0.001
C(2)-H(5) 1.095 1.092 -0.003
Here are the distances (Angstrom) as obtained with a QM and an Hybrid method with explicit typing:
Distance QM MM Err(MM)
-------------------------------------------
C(1)-C(2) 1.456 1.456 0.000
C(1)-N(4) 1.147 1.147 -0.001
C(2)-H(5) 1.095 1.092 -0.003
Here are some observations for this example:
* The hybrid engine does better than pure MM
* The subtle issue whether or not we specify the types has negligible effect
Note that this example makes use of the python scripting tools PLAMS and PISA, available in the AMS Python Stack.
#!/usr/bin/env amspython
from scm.base import ChemicalSystem
from scm.input_classes import drivers, engines
from scm.plams import AMSJob, JobRunner, config, Settings
config.default_jobrunner = JobRunner(parallel=True)
settings = Settings()
settings.runscript.nproc = 1
mol = ChemicalSystem(
"""
System
Atoms
C -0.02116 1.01286 0.0 region=qm
C 0.01258 -0.45034 0.0 region=qm
C 1.44394 -1.0175 0.0
N -0.03362 2.17616 0.0 region=qm
H -0.54281 -0.80179 0.88302 region=qm
H -0.54281 -0.80179 -0.88302 region=qm
H 1.40659 -2.11445 0.0
H 1.99584 -0.68766 -0.88907
H 1.99584 -0.68766 0.88907
End
GuessBonds true
End
"""
)
results = {}
for engine in [engines.DFTB(), engines.ForceField()]:
driver = drivers.AMS()
driver.Engine = engine
driver.Task = "GeometryOptimization"
job = AMSJob(molecule=mol, settings=settings + Settings({"input": driver}), name=engine.name)
results[driver.Engine.name] = job.run()
mol_qm = results["DFTB"].get_main_system()
mol_mm = results["ForceField"].get_main_system()
bonds = {"C(1)-C(2)": (0, 1), "C(1)-N(4)": (0, 3), "C(2)-H(5)": (1, 4)}
print("begin report")
print(
"""We first check how bad the MM method is compared to the QM method for some distances in the QM region.
Here are the distances (Angstrom) as obtained with a QM and an MM method:"""
)
header = "{:<10} {:>10} {:>10} {:>10}".format("Distance", "QM", "MM", "Err(MM)")
print(header)
print("-" * len(header))
for bond, (i, j) in bonds.items():
dist_qm = mol_qm.get_distance(i, j)
dist_mm = mol_mm.get_distance(i, j)
print(f"{bond:<10} {dist_qm:>10.3f} {dist_mm:>10.3f} {dist_mm - dist_qm:>10.3f}")
print(
"""Can we get better results for the QM region with the hybrid engine?
Even though UFF has automatic atom typing, it still matters (in principle) whether we specify it on input or not:
* Without typing for each region the types are automatically guessed
* With typing the types are always as on input (for all regions)
The only difference is in the C type for the MM region."""
)
results_typing = {}
for typing in [False, True]:
driver = drivers.AMS()
driver.Engine = engine
driver.Task = "GeometryOptimization"
driver.Engine = engines.Hybrid()
driver.Engine.QMMM.QMRegion = "qm"
driver.Engine.QMMM.QMEngineID = "DFTB"
driver.Engine.QMMM.MMEngineID = "ForceField"
driver.Engine.Engine[0] = engines.DFTB()
driver.Engine.Engine[1] = engines.ForceField()
if typing:
atom_types = results["ForceField"].get_atom_types()
mol.enable_atom_attributes("forcefield")
mm_atoms = mol.get_atoms_outside_region("qm")
mol.set_atoms_in_region(mm_atoms, "mm")
for atom, atom_type in zip(mol.atoms, atom_types):
atom.forcefield.type = atom_type
job = AMSJob(molecule=mol, settings=settings + Settings({"input": driver}), name=f"hybrid.types={typing}")
results_typing[typing] = job.run()
systems_typing = {k: v.get_main_system() for k, v in results_typing.items()}
for typing, system in systems_typing.items():
print(
f"Here are the distances (Angstrom) as obtained with a QM and an Hybrid method {'with' if typing else 'without'} explicit typing:"
)
header = "{:<10} {:>10} {:>10} {:>10}".format("Distance", "QM", "MM", "Err(MM)")
print(header)
print("-" * len(header))
for bond, (i, j) in bonds.items():
dist_qm = mol_qm.get_distance(i, j)
dist_hybrid = system.get_distance(i, j)
print(f"{bond:<10} {dist_qm:>10.3f} {dist_hybrid:>10.3f} {dist_hybrid - dist_qm:>10.3f}")
print(
"""Here are some observations for this example:
* The hybrid engine does better than pure MM
* The subtle issue whether or not we specify the types has negligible effect
"""
)
print("end report")