Examples¶
These examples show how to run Quantum ESPRESSO through the AMS driver.
All examples can be run from the command-line and most of them are also available in
$AMSHOME/examples/QEMost of the examples can also be copy-pasted or opened directly in the AMSinput GUI
Important
These examples do not necessarily contain scientifically good settings!
Typically you should increase the energy cutoff and k-point sampling for scientifically good results.
See also
Single-Point Calculation + Band Structure¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1 # must be set when using the Quantum ESPRESSO engine, see documentation
"$AMSBIN/ams" --delete-old-results << EOF
Task SinglePoint
System
Atoms
Si -0.67520366 -0.67520366 -0.67520366
Si 0.67520366 0.67520366 0.67520366
End
Lattice
0.00000000 2.70081465 2.70081465
2.70081465 0.00000000 2.70081465
2.70081465 2.70081465 0.00000000
End
End
Engine QuantumEspresso
System
ecutwfc 50
ecutrho 400
occupations smearing
degauss 0.001
End
K_Points automatic
5 5 5 0 0 0
End
Pseudopotentials
Family pslibrary-US
Functional PBE
End
Properties
BandStructure Yes
End
EndEngine
EOF
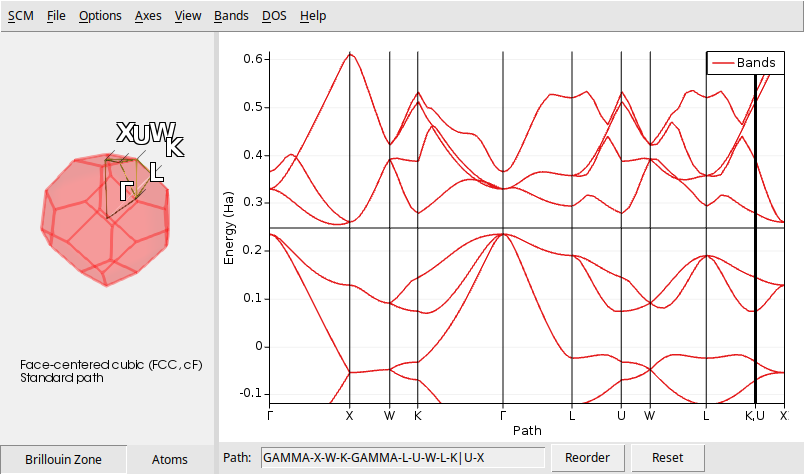
Single-point calculation for bulk silicon using a regular Monkhorst-Pack k-point grid.
The electronic band structure is calculated along high-symmetry lines in the Brillouin zone.
You can view the resulting band structure with the program
$AMSBIN/amsbands.
Lattice Optimization of Silicon¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1 # required for Quantum ESPRESSO - see documentation
#============================================================================
# Example showing how to run a lattice optimization with Quantum ESPRESSO.
#
# This example is also available as a tutorial using the
# graphical user interface
#
# Important: always use carefully converged k-points and energy cutoffs
# for lattice optimizations!
#
# You may want to symmetrize the optimized structure using the "star"
# button in AMSinput.
#============================================================================
"$AMSBIN/ams" --delete-old-results <<EOF
Task GeometryOptimization
GeometryOptimization
OptimizeLattice Yes
End
System
Atoms
Si -0.67875000 -0.67875000 -0.67875000
Si 0.67875000 0.67875000 0.67875000
End
Lattice
0.0 3.0 3.0
3.0 0.0 3.0
3.0 3.0 0.0
End
End
Engine QuantumEspresso
K_Points automatic
10 10 10 0 0 0
End
Pseudopotentials
Family SSSP-Efficiency
Functional PBE
End
System
ecutwfc 40.0
ecutrho 320.0
occupations Fixed
End
EndEngine
EOF
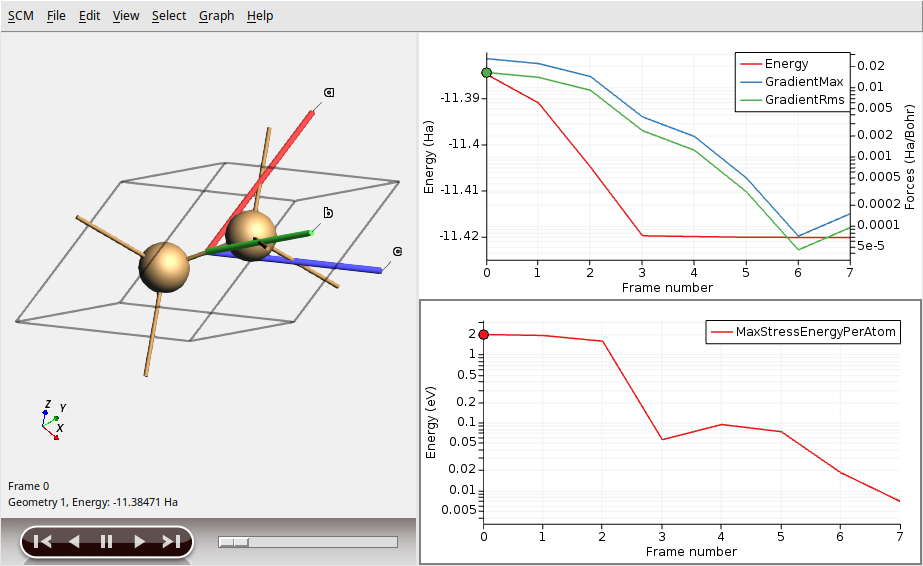
Full structural optimization of bulk silicon
Both the atomic positions and lattice parameters are relaxed
DFT+U (Hubbard U) Calculation for Anti-ferromagnetic FeO¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
# Run AMS driver without MPI, so that QE can be MPI parallel.
export SCM_DISABLE_MPI=1
"$AMSBIN/ams" --delete-old-results << EOF
Task SinglePoint
System
Atoms
Fe 0.0 0.0 0.0 QE.Label=Fe1
Fe 2.165 2.165 0.0 QE.Label=Fe2
O 0.0 2.165 0.0
O 2.165 0.0 0.0
End
Lattice
4.33 0.0 0.0
0.0 4.33 0.0
2.165 0.0 2.165
End
End
Engine QuantumEspresso
System
ecutwfc 40.0
ecutrho 160.0
occupations smearing
smearing mv
degauss 0.02
nspin 2
starting_magnetization label=Fe1 value=0.5
starting_magnetization label=Fe2 value=-0.5
End
Pseudopotentials
Family pslibrary-PAW
Functional PBEsol
End
K_Points automatic
5 5 5 0 0 0
End
Hubbard ortho-atomic
U Fe1-3d 4.6
U Fe2-3d 4.6
End
EndEngine
EOF
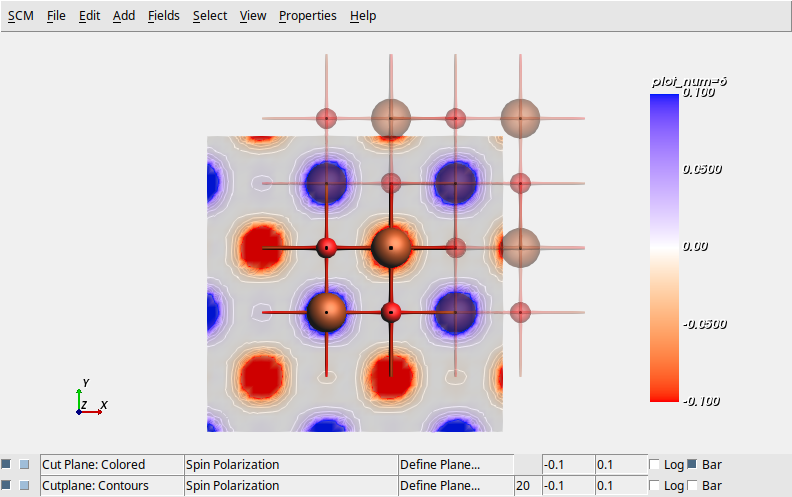
DFT+U calculation for antiferromagnetic FeO
The
QE.Label=keyword is employed to define distinct Fe speciesThe
starting_magnetizationkeyword refers to the labels to give different initial spin magnetizations, leading to an anti-ferromagnetic solution.Hubbard U is specified in the new Quantum ESPRESSO 7.1 format
Pseudopotential Selection¶
Command-line runscript
#!/bin/sh
export SCM_DISABLE_MPI=1 # required for engine Quantum ESPRESSO, see documentation
# =======================================================================================
# Example illustrating various ways of selecting Pseudopotentials with the QE engine.
# =======================================================================================
#
# All jobs run into this example should give the exact same result,
# as they are using the same PPs.
# All that differs is the way that the PPs were selected.
# Make a folder with "custom" PP files to test with:
#
# We will use the SG15 pseudopotentials directory that comes
# with the AMS QE package as an example.
# You could use any other directory with .upf files ...
PPDIR="$("$AMSBIN/amspackages" loc qe)/upf_files/GGA/PBE/SR/SG15-1.2/UPFs"
# Make a custom directory containing files named element.upf:
mkdir -p my_pp_directory
cp "$PPDIR"/C_ONCV_PBE-1.2.upf my_pp_directory/C.upf # rename to element.upf
cp "$PPDIR"/H_ONCV_PBE-1.2.upf my_pp_directory/H.upf # rename to element.upf
# If using special QE labels, the file name should be label.upf:
cp "$PPDIR"/H_ONCV_PBE-1.2.upf my_pp_directory/H1.upf
# Get an absolute path to the directory the jobs starts in:
if test "$OS" = "Windows_NT"; then
# Absolute paths should be Windows style (with drive letters like C:)
STARTDIR=$(pwd -W)
else
STARTDIR=$(pwd)
fi
# Make a system to test with:
echo "
System
Atoms
C 0.9685 1.9663 0.0000
H 1.6614 2.2162 0.7898
H 0.0162 1.6927 0.4297
H 0.8388 2.8201 -0.6485
H 1.3576 1.1363 -0.5710 QE.Label=H1
End
Lattice
5.0 0.0 0.0
0.0 5.0 0.0
0.0 0.0 5.0
End
End
" > system.in
# Option 1: Pseudopotentials%Family
# ---------------------------------
#
# The easiest way is to just use the Pseudopotentials%Family key and
# let the engine figure out which PPs to use.
AMS_JOBNAME=1_family "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Family SG15
Functional PBE
End
K_points Gamma
End
EndEngine
EOF
# Option 2: Pseudopotentials%Directory
# ------------------------------------
#
# Specifies a path to a directory with PP files named element.upf or label.upf.
# The path to the directory can be absolute or relative.
AMS_JOBNAME=2a_directory_relpath "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Directory my_pp_directory
End
K_points Gamma
End
EndEngine
EOF
AMS_JOBNAME=2b_directory_abspath "$AMSBIN/ams" << EOF
Task SinglePoint
System
Atoms
C 0.9685 1.9663 0.0000 QE.Label=C1 # no PP file for C1 in folder, fallback to C
H 1.6614 2.2162 0.7898
H 0.0162 1.6927 0.4297
H 0.8388 2.8201 -0.6485
H 1.3576 1.1363 -0.5710 QE.Label=H1
End
Lattice
5.0 0.0 0.0
0.0 5.0 0.0
0.0 0.0 5.0
End
End
Engine QuantumEspresso
Pseudopotentials
Directory $STARTDIR/my_pp_directory
End
K_points Gamma
End
EndEngine
EOF
# Option 3: Pseudopotentials%Files
# ------------------------------------
#
# Individually specify paths to files for each element or label.
# By default relative paths are relative to the root of the
# PP library installed via AMSpackages.
AMS_JOBNAME=3a_files_relpath "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Files Label=C Path=GGA/PBE/SR/SG15-1.2/UPFs/C_ONCV_PBE-1.2.upf
Files Label=H Path=GGA/PBE/SR/SG15-1.2/UPFs/H_ONCV_PBE-1.2.upf
Files Label=H1 Path=GGA/PBE/SR/SG15-1.2/UPFs/H_ONCV_PBE-1.2.upf
End
K_points Gamma
End
EndEngine
EOF
# Paths relative to the starting directory of AMS should be prefixed with "./".
AMS_JOBNAME=3b_files_exrelpath "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Files Label=C Path=./my_pp_directory/C.upf
Files Label=H Path=./my_pp_directory/H.upf
Files Label=H1 Path=./my_pp_directory/H1.upf
End
K_points Gamma
End
EndEngine
EOF
# Absolute paths are also supported.
AMS_JOBNAME=3c_files_abspath "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Files Label=C Path="$STARTDIR/my_pp_directory/C.upf"
Files Label=H Path="$STARTDIR/my_pp_directory/H.upf"
Files Label=H1 Path="$STARTDIR/my_pp_directory/H1.upf"
End
K_points Gamma
End
EndEngine
EOF
# Combinations between absolute, relative and explicit relative paths also work.
AMS_JOBNAME=3d_files_mixed "$AMSBIN/ams" << EOF
Task SinglePoint
@include system.in
Engine QuantumEspresso
Pseudopotentials
Files Label=C Path=GGA/PBE/SR/SG15-1.2/UPFs/C_ONCV_PBE-1.2.upf
Files Label=H Path=./my_pp_directory/H.upf
Files Label=H1 Path="$STARTDIR/my_pp_directory/H1.upf"
End
K_points Gamma
End
EndEngine
EOF
This example illustrates various methods for selecting pseudopotentials. It covers:
Pseudopotentials%Family: Predefined sets for simple selection.Pseudopotentials%Directory: Directory containing filesH.upf,He.upf, etc.Pseudopotentials%Files: Specify each individual pseudopotential file (similar to standalone Quantum ESPRESSO).
All these options are also accessible from the AMSinput GUI.
Slab With Dipole Correction, Work Function¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
export AMS_JOBNAME=DipoleCorrection_WorkFunction
#============================================================================
# Example showing how to apply a dipole correction for a slab system.
# This gives two separate vacuum levels on the different sides of the slab.
# The respective work functions are calculated as differences
# between the vacuum levels and the Fermi energy.
#
# This example uses internally the pp.x and average.x utilities from Quantum
# ESPRESSO to get the plane-averaged electrostatic potential.
#
# For details about the input to pp.x, and average.x, see:
# - https://www.quantum-espresso.org/Doc/INPUT_PP.html
# - https://gitlab.com/QEF/q-e/-/blob/qe-7.1/PP/src/average.f90
#
# This example is based on the work function tutorial for BAND and the
# work function example in Quantum ESPRESSO (PP/examples/WorkFct_example).
#
# Better-converged numbers can be obtained by increasing the k-space
# sampling and energy cutoff.
#============================================================================
"$AMSBIN/ams" --delete-old-results <<EOF
Task SinglePoint
System
Atoms
Al 1.4319 1.4319 8.925
Al 0.0000 0.0000 10.95
Al 1.4319 1.4319 12.975
Al 0.0000 0.0000 15
F 0.0000 0.0000 18.27
Li 1.4319 1.4319 18.27
F 1.4319 1.4319 20.295
Li 0.0000 0.0000 20.295
End
Lattice
2.86378246 0.00000000 0.00000000
0.00000000 2.86378246 0.00000000
0.0 0.0 30.0
End
End
Engine QuantumEspresso
K_Points automatic
2 2 1 1 1 1
End
Pseudopotentials
Family GBRV
Functional PBE
End
System
ecutwfc 40.0
occupations smearing
smearing gaussian
degauss 0.02
edir 3
emaxpos 0.90
eopreg 0.10
End
Electrons
conv_thr 1.0e-10
End
Control
tefield Yes
dipfield Yes
End
Properties
WorkFunction Yes
End
EndEngine
EOF
$AMSBIN/amspython -c '
import scm.plams
import matplotlib.pyplot as plt
job = scm.plams.AMSJob.load_external("'$AMS_JOBNAME'.results/ams.rkf")
wf_results = job.results.get_work_function_results( "eV", "Angstrom" )
ax = scm.plams.plot_work_function(*wf_results)
ax.set_title("Electrostatic Potential Profile", fontsize=14)
ax.set_xlabel("Shifted coordinate (angstrom)", fontsize=13)
ax.set_ylabel("Electrostatic potential (eV)", fontsize=13)
plt.savefig("work_function.png")
'
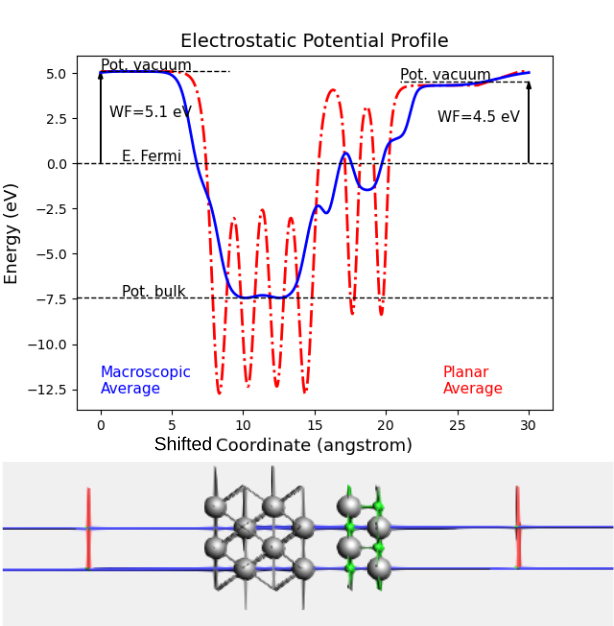
Recommended: Place the system in the middle of the unit cell to calculate the work function.
Apply a dipole correction to account for the dipole moment across the vacuum between periodic images
This gives two separate vacuum levels on the different sides of the slab
The work functions are calculated as the difference between the vacuum level and the Fermi energy.
Numerical Calculation of Phonons¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
AMS_JOBNAME=DiamondNumericalPhonons_Curtarolo "$AMSBIN/ams" --delete-old-results << EOF
Task SinglePoint
System
Atoms
C -0.44625000 -0.44625000 -0.44625000
C 0.44625000 0.44625000 0.44625000
End
Lattice
0.00000000 1.78500000 1.78500000
1.78500000 0.00000000 1.78500000
1.78500000 1.78500000 0.00000000
End
End
Properties
Phonons Yes
End
Phonons
Method Numerical
End
NumericalPhonons
SuperCell
2 0 0
0 2 0
0 0 2
End
AutomaticBZPath Yes
End
Engine QuantumEspresso
K_Points automatic
3 3 3 0 0 0
End
EndEngine
EOF
# This section uses PLAMS to plot the phonons bands in a Python environment
AMS_JOBNAME=DiamondNumericalPhonons_Curtarolo $AMSBIN/amspython -c '
import os
import scm.plams
import matplotlib.pyplot as plt
results_file = os.environ["AMS_JOBNAME"]+".results/ams.rkf"
job = scm.plams.AMSJob.load_external(results_file)
phonons_data = job.results.get_phonons_band_structure(unit="cm^-1")
ax = scm.plams.plot_phonons_band_structure(*phonons_data)
ax.set_ylabel("$E$ (cm$^{-1}$)")
ax.set_xlabel("Path")
ax.set_title("Diamond Phonons")
plt.savefig("phonons.png")
properties_data = job.results.get_phonons_thermodynamic_properties( properties_unit=["kJ/mol", "J/K/mol"] )
ax = scm.plams.plot_phonons_thermodynamic_properties(*properties_data)
ax.set_ylabel("Thermodynamic Properties")
ax.set_xlabel("Temperature (K)")
plt.savefig("thermodynamics.png")
'
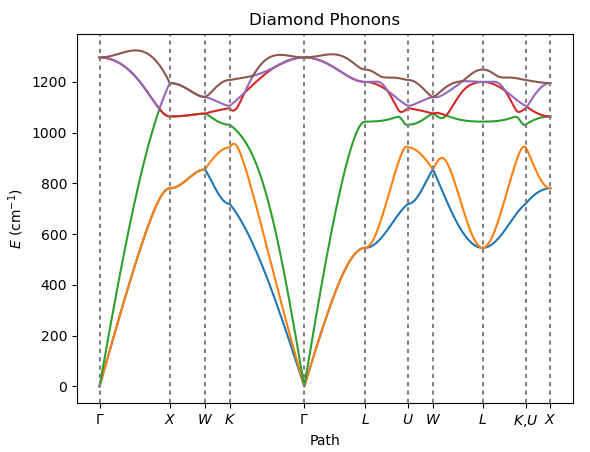
Calculate phonon dispersion curves of diamond using numerical differentiation.
The dynamical matrix is constructed within the supercell approximation by computing forces resulting from finite atomic displacements.
Visualize the results with the AMSbands GUI or the Python PLAMS library.
Analytical Calculation of Phonons Using DFPT¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
AMS_JOBNAME=DiamondPhonons_Curtarolo "$AMSBIN/ams" --delete-old-results << EOF
Task SinglePoint
System
Atoms
C -0.44625000 -0.44625000 -0.44625000
C 0.44625000 0.44625000 0.44625000
End
Lattice
0.00000000 1.78500000 1.78500000
1.78500000 0.00000000 1.78500000
1.78500000 1.78500000 0.00000000
End
End
Properties
Phonons Yes
End
Engine QuantumEspresso
K_Points automatic
3 3 3 0 0 0
End
Phonons
asr crystal
Q_Points automatic
4 4 4 0 0 0
End
End
EndEngine
EOF
# This section uses PLAMS to plot the phonons bands in a Python environment
AMS_JOBNAME=DiamondPhonons_Curtarolo $AMSBIN/amspython -c '
import os
import scm.plams
import matplotlib.pyplot as plt
results_file = os.environ["AMS_JOBNAME"]+".results/ams.rkf"
job = scm.plams.AMSJob.load_external(results_file)
phonons_data = job.results.get_phonons_band_structure(unit="cm^-1")
ax = scm.plams.plot_phonons_band_structure(*phonons_data)
ax.set_ylabel("$E$ (cm$^{-1}$)")
ax.set_xlabel("Path")
ax.set_title("Diamond Phonons")
plt.savefig("phonons.png")
properties_data = job.results.get_phonons_thermodynamic_properties( properties_unit=["kJ/mol", "J/K/mol"] )
ax = scm.plams.plot_phonons_thermodynamic_properties(*properties_data)
ax.set_ylabel("Thermodynamic Properties")
ax.set_xlabel("Temperature (K)")
plt.savefig("thermodynamics.png")
'
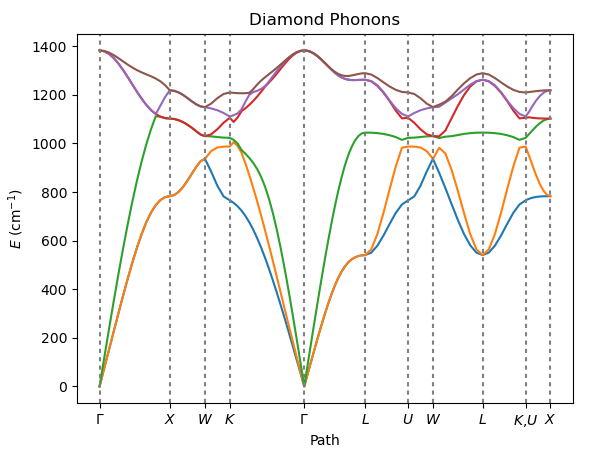
Calculate phonon dispersion curves of diamond using Density Functional Perturbation Theory (DFPT) as implemented in Quantum ESPRESSO.
DFPT allows for the analytical computation of the dynamical matrix.
Visualize the results with the AMSbands GUI or the Python PLAMS library.
IR/Raman Spectra¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
export AMS_JOBNAME=BeO
"$AMSBIN/ams" --delete-old-results << EOF
Task GeometryOptimization
Properties
NormalModes Yes
Raman Yes
End
System
Atoms
Be 0.0000000000 1.5555413800 0.0007009000
Be 1.3471383600 0.7777706900 2.1887103500
O 0.0000000000 1.5555413800 1.6512462200
O 1.3471383600 0.7777706900 3.8392556700
End
Lattice
2.6942767100 0.0000000000 0.0000000000
-1.3471383600 2.3333120800 0.0000000000
0.0000000000 0.0000000000 4.3760188900
End
End
Engine QuantumEspresso
System
ecutwfc 25.0
End
K_Points automatic
4 4 4 0 0 0
End
Pseudopotentials
Files Label=Be Path=QE/Be.pz-mt_fhi.UPF
Files Label=O Path=QE/O.pz-mt_fhi.UPF
End
EndEngine
EOF
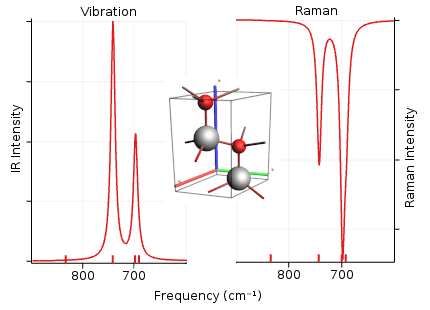
Calculate the infrared and Raman spectra of BeO using DFPT
Use norm-conserving pseudopotentials
Raman only available for LDA
Visualize the calculated IR and Raman spectra, and also the corresponding vibrational modes with the program
$AMSBIN/amsspectra
PESScan: volume scan¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
"$AMSBIN/ams" --delete-old-results <<EOF
Task PESScan
System
Atoms
Mg 0.000 0.000 0.000
O 2.105 2.105 2.105
End
Lattice
0.000 2.105 2.105
2.105 0.000 2.105
2.105 2.105 0.000
End
End
PESScan
CalcPropertiesAtPESPoints Yes
ScanCoordinate
nPoints 5
CellVolumeScalingRange 0.9 1.2
End
End
Engine QuantumEspresso
K_Points automatic
2 2 2 0 0 0
End
Properties
DOS Yes
BandStructure Yes
End
DOS
K_Points automatic
10 10 10 0 0 0
End
End
BandStructure
K_Points ams_kpath
End
End
EndEngine
EOF
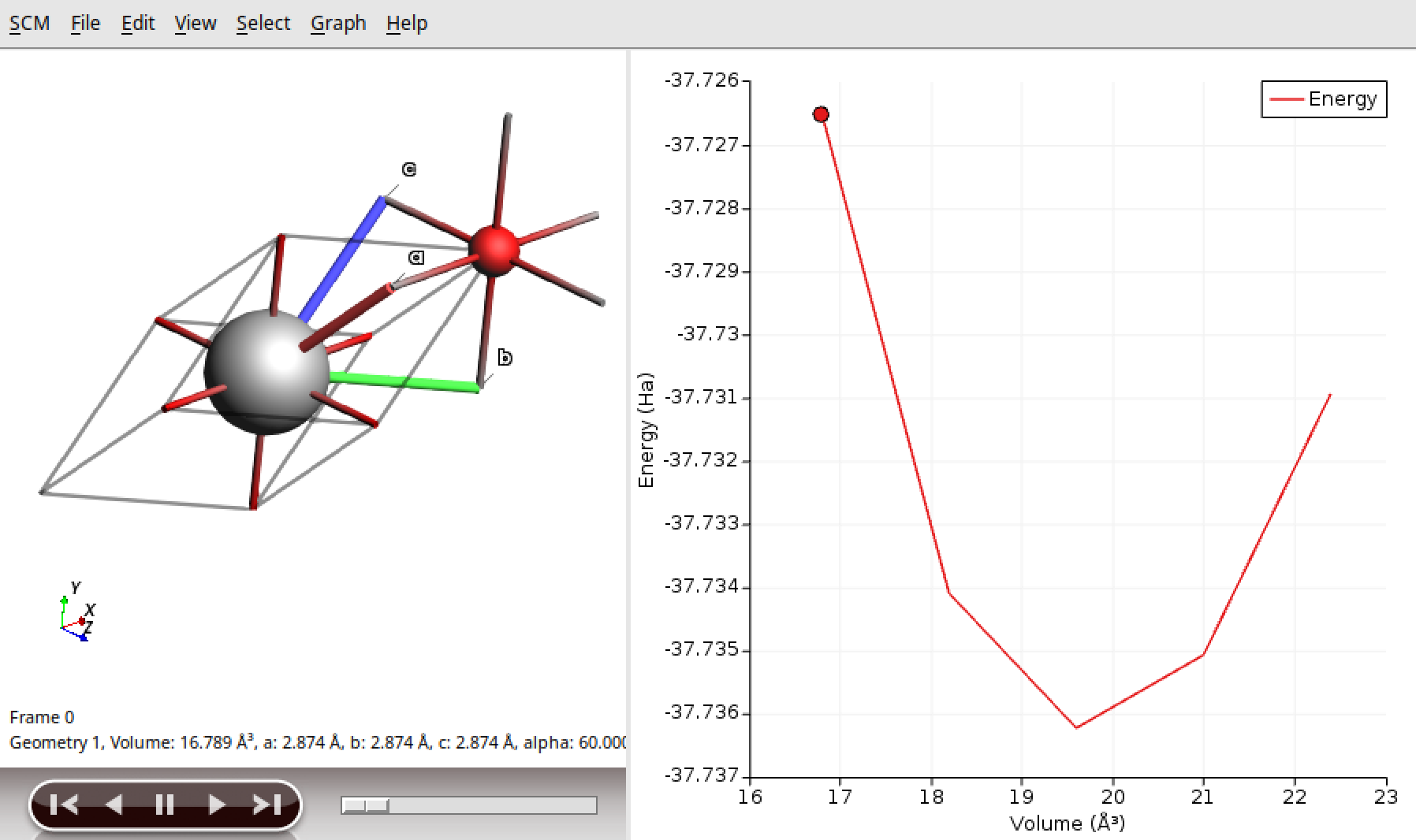
Use the
PESScantask in combination with the Quantum ESPRESSO engine. It performs a series of geometry optimization calculations while systematically varying the cell volume of MgO.The density of states (DOS) and band structure are calculated for each volume.
You can analyze the results with the program
$AMSBIN/amsmovie.
Replay¶
Command-line runscript
#!/bin/sh
export SCM_DISABLE_MPI=1
AMS_JOBNAME=PESScan "$AMSBIN/ams" --delete-old-results <<EOF
Task PESScan
System
Atoms
Mg 0.000 0.000 0.000
O 2.105 2.105 2.105
End
Lattice
0.000 2.105 2.105
2.105 0.000 2.105
2.105 2.105 0.000
End
End
PESScan
ScanCoordinate
nPoints 3
CellVolumeScalingRange 0.9 1.2
End
End
Engine QuantumEspresso
K_Points automatic
2 2 2 0 0 0
End
EndEngine
EOF
AMS_JOBNAME=Replay "$AMSBIN/ams" --delete-old-results << EOF
Task Replay
Replay
File PESScan.results/ams.rkf
End
Properties
Gradients Yes
StressTensor Yes
End
Engine QuantumEspresso
K_Points automatic
5 5 5 0 0 0
End
EndEngine
EOF
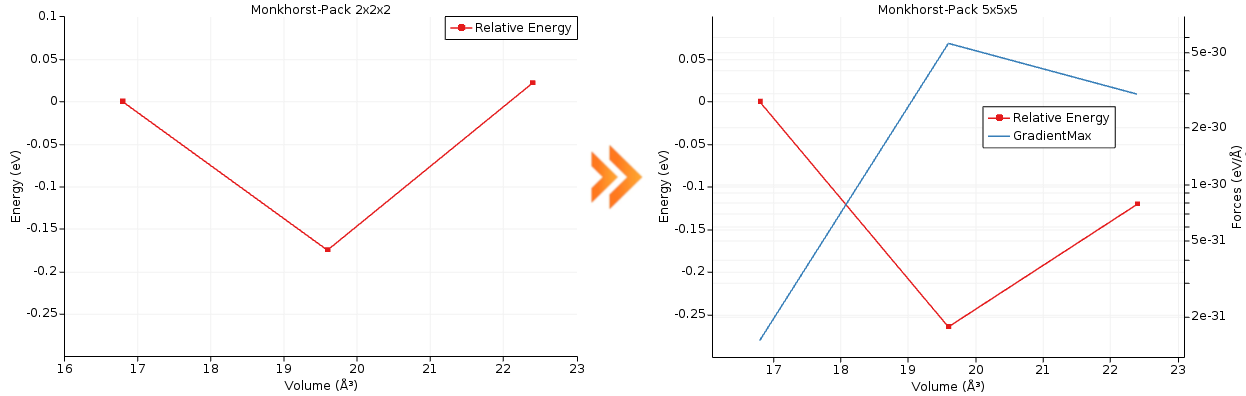
This example demonstrates a two-step approach for efficient property calculations. Initially, a PESScan task is performed with a low-precision setup (e.g., coarse k-point grid) to quickly generate a series of structures, using a cell volume scaling for MgO as a prototype system. Then, the Replay task is used to evaluate properties like gradients and stress tensors at a higher precision (e.g., finer k-point grid) for the selected structures.
Born-Oppenheimer Molecular Dynamics¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
AMS_JOBNAME=MolecularDynamics "$AMSBIN/ams" --delete-old-results << EOF
Task MolecularDynamics
RNGSeed 10
MolecularDynamics
NSteps 10
Trajectory
SamplingFreq 1
EngineResultsFreq 0
End
Checkpoint
Frequency 1
End
InitialVelocities
Temperature 300
End
End
System
Atoms
O 0.77656125 0.70226469 -0.77898886
H 0.18695701 1.27832345 -0.18188342
H 1.36427986 1.27137397 -1.38443744
O -0.77660318 -0.83048394 0.77894720
H -0.18661066 -0.26385475 1.38473471
H -1.36396069 -0.25091023 0.18224739
End
Lattice
3.10979012 0.00175445 -0.000372438
-0.00721068 3.06655157 -0.007924867
-3.8697e-05 0.00424969 3.111852930
End
End
Engine QuantumEspresso
EndEngine
EOF
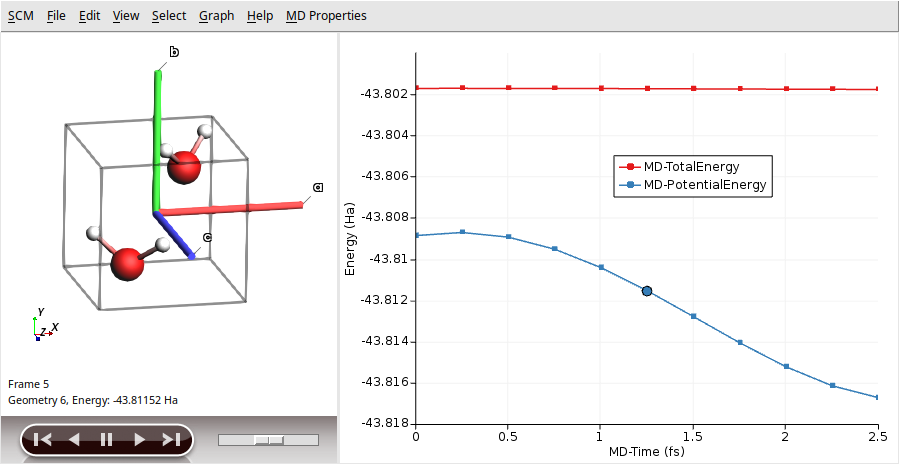
Born-Oppenheimer Molecular Dynamics (BOMD) simulation with the AMS Driver
Analyze the resulting trajectory with the program
$AMSBIN/amsmovie
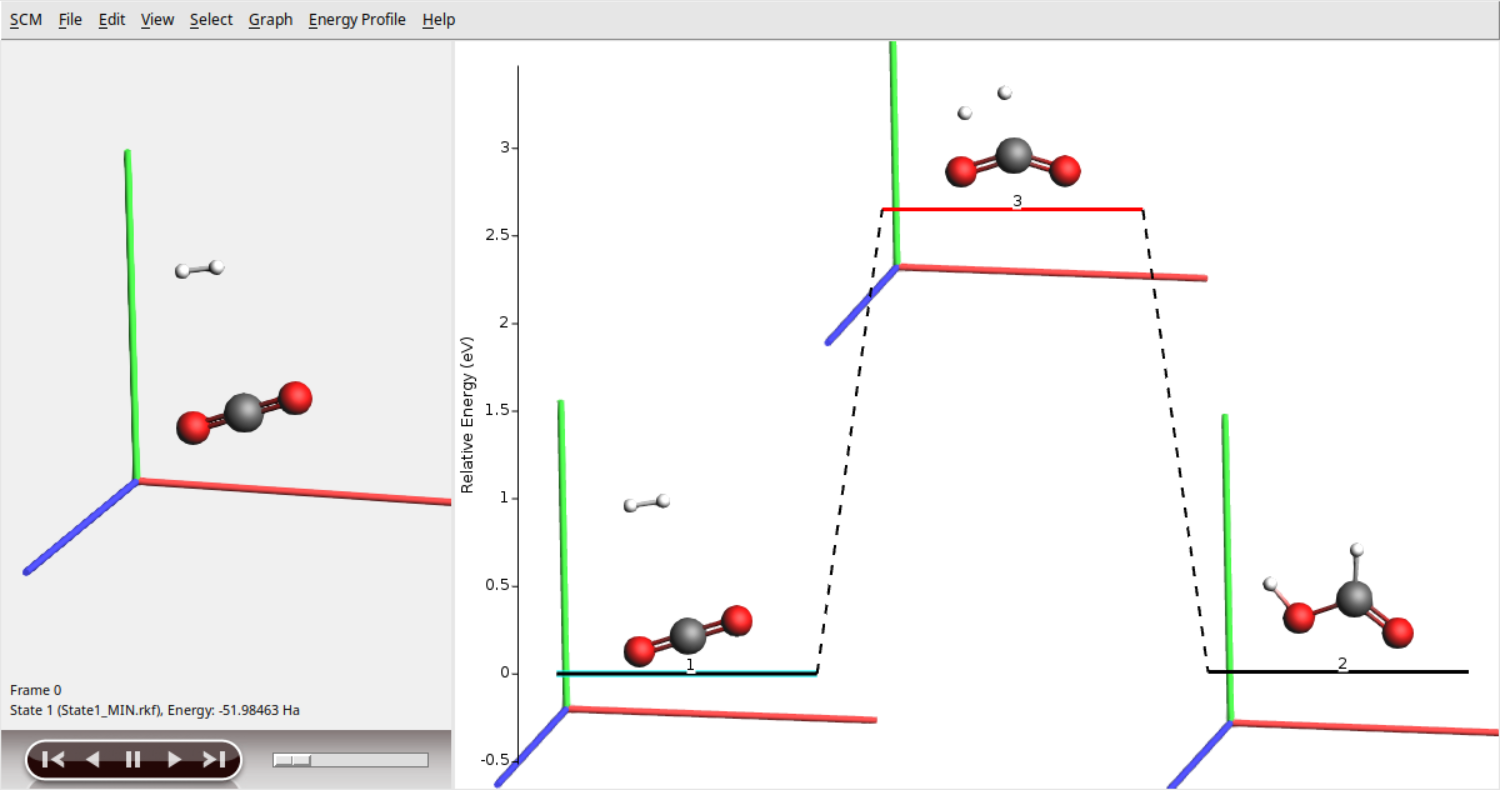
PES Exploration¶
Input File (run from command-line or the AMSinput GUI)
#!/bin/sh
export SCM_DISABLE_MPI=1
"$AMSBIN/ams" --delete-old-results << EOF
Task PESExploration
PESExploration
Job SaddleSearch
RandomSeed 10
Optimizer
ConvergedForce 0.05
End
SaddleSearch
DisplaceMagnitude 0.01
RelaxFromSaddlePoint Yes
End
End
System
Atoms
O 3.63620880 3.57049155 4.48487828
C 4.48799778 4.50659100 4.51176734
O 5.64352871 4.78595471 4.54849633
H 3.09645968 4.80015429 4.45130367
H 3.65975624 5.66218780 4.46883580
End
Lattice
9.0 0.0 0.0
0.0 9.0 0.0
0.0 0.0 9.0
End
End
Engine QuantumEspresso
System
ecutwfc 30.0
ecutrho 120.0
assume_isolated Martyna-Tuckerman
End
K_Points automatic
1 1 1 0 0 0
End
NormalModes
asr crystal
End
EndEngine
EOF
This example demonstrates the use of the Quantum ESPRESSO engine in combination with the PESExploration task. The initial structure is a guess to the transition state of the reaction \(H_2+CO_2 \longrightarrow HOCHO\). Here, AMS locates the actual transition state and the corresponding reactants and products in an automated fashion.