PES point properties¶
No matter what application the AMS driver is used for, in one way or another it
always explores the potential energy surface (PES) of the system. One can
furthermore ask AMS to calculate additional properties of the PES in the points
that are visited. These are mostly (but not exclusively) derivatives of the energy, e.g. we can ask
AMS to calculate the gradients or the Hessian in the visited points. In general
all these PES point properties are requested through the Properties block in
the AMS input:
Properties
Gradients [True | False]
StressTensor [True | False]
Hessian [True | False]
SelectedAtomsForHessian integer_list
NormalModes [True | False]
PESPointCharacter [True | False]
ElasticTensor [True | False]
Phonons [True | False]
DipoleMoment [True | False]
BondOrders [True | False]
End
This page in the AMS manual describes all the supported properties.
Note that because these properties are tied to a particular point on the potential energy surface, they are found on the engine output files. Note also that the properties are not always calculated in every PES point that the AMS driver visits during a calculation. By default they are only calculated in special PES points, where the definition of special depends on the task AMS is performing: For a geometry optimization properties would for example only be calculated at the final, converged geometry. This behavior can often be modified by keywords special to the particular running task.
Nuclear gradients and stress tensor¶
The first derivative with respect to the nuclear coordinates can be requested as follows:
Properties
Gradients [True | False]
End
PropertiesGradientsType: Bool Default value: False GUI name: Nuclear gradients Description: Calculate the nuclear gradients.
Note that these are gradients, not forces, the difference being the sign. The
gradients are printed in the output and written to the engine result
file belonging to the particular point on the PES in the
AMSResults%Gradients variable as a \(3 \times n_\mathrm{atoms}\) array
in atomic units (Hartree/Bohr).
For periodic systems (chains, slabs, bulk) one can also request the clamped-ion stress tensor (note: the clamped-ion stress is only part of the true stress tensor):
Properties
StressTensor [True | False]
End
PropertiesStressTensorType: Bool Default value: False GUI name: Stress tensor Description: Calculate the stress tensor.
The clamped-ion stress tensor \(\sigma_\alpha\) (Voigt notation) is computed via numerical differentiation of the energy \(E\) WRT a strain deformations \(\epsilon_\alpha\) keeping the atomic fractional coordinates constant:
where \(V_0\) is the volume of the unit cell (for 2D periodic system \(V_0\) is the area of the unit cell, and for 1D periodic system \(V_0\) is the length of the unit cell).
The clamped-ion stress tensor (in Cartesian notation) is written to the engine
result file in AMSResults%StressTensor.
Hessian and normal modes of vibration¶
The calculation of the second derivative of the total energy with respect to the nuclear coordinates is enabled by:
Properties
Hessian [True | False]
End
PropertiesHessianType: Bool Default value: False Description: Whether or not to calculate the Hessian.
The Hessian is not printed to the text output, but is saved in the engine result
file as variable AMSResults%Hessian. Note that this ist just the plain
second derivative (no mass-weighting) of the total energy and that for the order
of its \(3 \times n_\mathrm{atoms}\) columns/rows, the component index
increases the quickest: The first column refers to changes in the
\(x\)-component of atom 1, the second to the \(y\)-component, the
fourth to the \(x\)-component of the second atoms, and so on.
It is also possible to request the calculation of the normal modes of vibration:
Properties
NormalModes [True | False]
End
PropertiesNormalModesType: Bool Default value: False GUI name: Frequencies Description: Calculate the frequencies and normal modes of vibration, and for molecules also the corresponding IR intensities.
Note
For more information and advanced methods of calculating and analyzing molecular vibrations, see the manual chapter on the vibrational analysis (mode scanning, refinement and tracking).
This implies the calculation of the Hessian, which is required for calculating
normal modes. For engines that are capable of calculating dipole moments, this
also enables the calculation of the infrared intensities, so that the IR
spectrum can be visualized by opening the engine result file with ADFSpectra.
The normal modes of vibration and the IR intensities are saved to the
engine result file in the Vibrations section.
Note
The calculation of the normal modes of vibration needs to be done the
system’s equilibrium geometry. So one should either run the normal modes
calculation using an already optimized geometry, or combine both steps into
one job by using the geometry optimization task
together with the Properties%NormalModes keyword.
Symmetry labels of the normal modes of molecules may be calculated if AMS uses symmetry in the calculation of normal modes (key NormalModes%UseSymmetry).
If symmetry is used the nomal modes are projected against symmtric displacements for each irrep. If that is not successful the symmetric label is ‘MIX’.
Symmetry is only recognized if the starting geometry has symmetry.
Symmetry is only used for molecules if the molecule has a specific orientation in space, like that the z-axis is the main rotation axis.
If the GUI is used one can click the Symmetrize button (the star), such that the GUI will (try to) symmetrize and reorient the molecule.
There are some cases that even after such symmetrization, the orientation of the molecule is not what is needed for the symmetry to be used.
If that is the case or if key NormalModes%UseSymmetry is set to ‘False’ or if there is no symmetry, then no symmetry labels will be calculated.
NormalModes
UseSymmetry [True | False]
End
NormalModesType: Block Description: Configures details of a normal modes calculation. UseSymmetryType: Bool Default value: True Description: Whether or not to exploit the symmetry of the system in the normal modes calculation.
The user may enable the ScanModes keyword to recalculate specific modes
after a normal modes calculation. It is identical to the ScanFreq option
that was available for older versions of ADF and BAND. For more information
on its use and purpose, see the Vibrational Analysis
documentation.
NormalModes
ScanModes [True | False]
FreqRange float_list
End
NormalModesScanModesType: Bool Default value: False Description: Whether or not to scan imaginary modes after normal modes calculation has concluded. FreqRangeType: Float List Default value: [-10000000.0, 10.0] Unit: cm-1 Recurring: True Description: Specifies a frequency range within which all modes will be scanned. (2 numbers: an upper and a lower bound.)
Thermodynamics (ideal gas)¶
The following thermodynamic properties are calculated by default whenever normal modes are computed: entropy, internal energy, constant volume heat capacity, enthalpy and Gibbs free energy. Translational, rotational and vibrational contributions are calculated for entropy, internal energy and constant volume heat capacity. Moments of Inertia and principal axis of the system are also computed. These results are written to the output file (section: “Statistical Thermal Analysis”) and to the engine binary results file (section: “Thermodynamics”).
The thermodynamic properties are computed assuming an ideal gas, and electronic contributions are ignored. The latter is a serious omission if the electronic configuration is (almost) degenerate, but the effect is small whenever the energy difference with the next state is large compared to the vibrational frequencies. The thermal analysis is based on the temperature dependent partition function. The energy of a (non-linear) molecule is (if the energy is measured from the zero-point energy)
The summation is over all harmonic \(\nu_j\), \(h\) is Planck’s constant and \(D\) is the dissociation energy
Contributions from low (less than 20 1/cm) frequencies to entropy, heat capacity and internal energy are excluded from the total values, but they are listed separately (so the user can add them if they wish).
Thermo
Pressure float
Temperatures float_list
End
ThermoType: Block Description: Options for thermodynamic properties (assuming an ideal gas). The properties are computed for all specified temperatures. PressureType: Float Default value: 1.0 Unit: atm Description: The pressure at which the thermodynamic properties are computed. TemperaturesType: Float List Default value: [298.15] Unit: Kelvin Description: List of temperatures at which the thermodynamic properties will be calculated.
Partial Vibrational Spectra (PVDOS)¶
The Partial Vibrational Spectra (also known as PVDOS) is computed by default whenever normal modes are requested. The PVDOS \(P_{I,n}\) for atom \(I\) and normal mode \(n\) is defined as:
where \(m_I\) is the nuclear weight of atom \(I\), and \(\vec{\eta}_{I,n}\) is the displacement vector for atom \(I\) in normal normal mode \(n\).
Tip
The Partial Vibrational Spectra (PVDOS) can be visualized using the ADFSpectra GUI module (Vibrations → Partial Vibrational Spectra (PVDOS)). When plotting a partial vibrational spectrum, the IR intensity of normal modes is scaled by the corresponding PVDOS of the selected atoms.
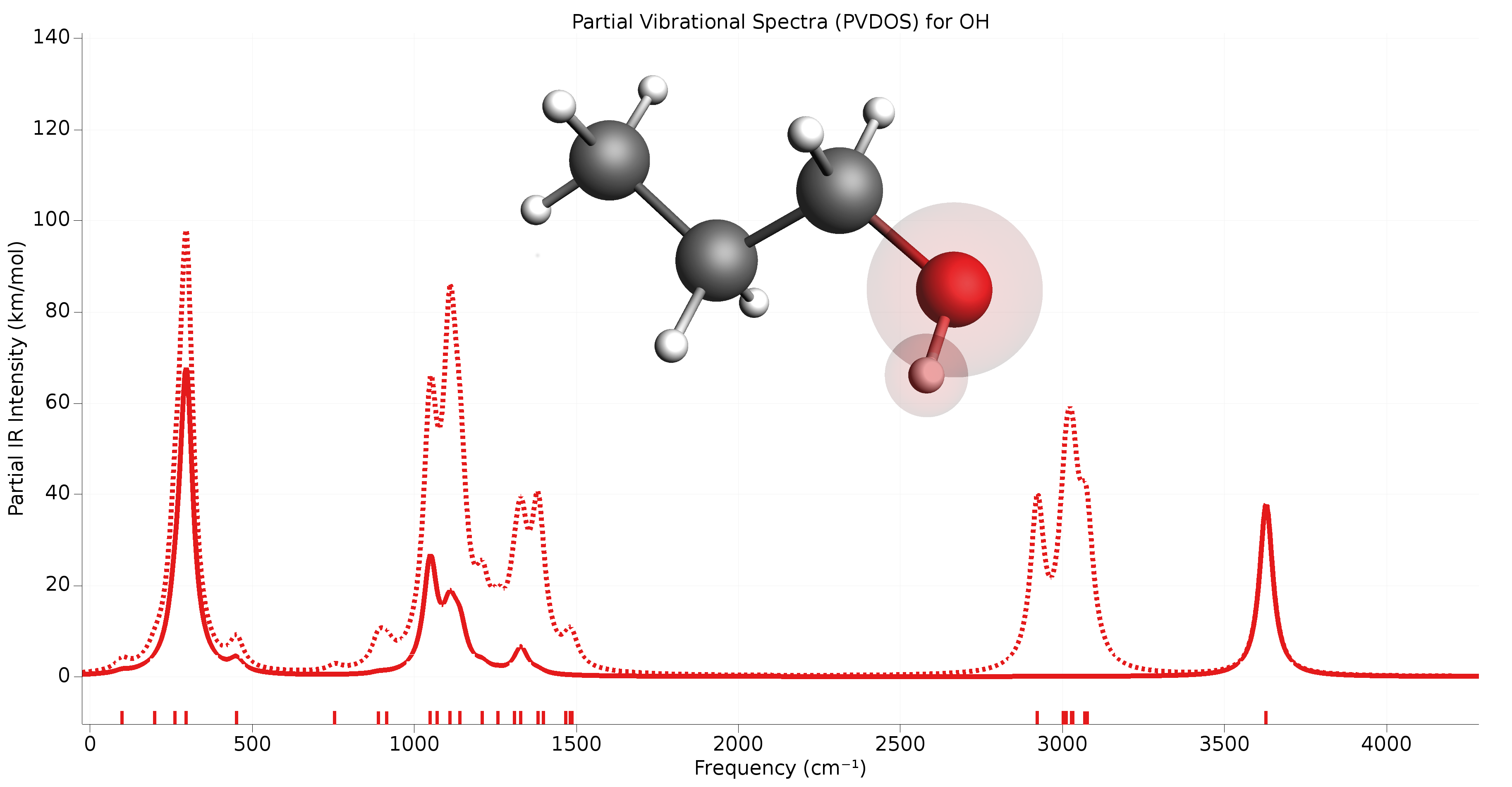
Fig. 2 Example of partial vibrational spectrum (PVDOS). The dotted line is the full IR spectrum of 1-propanol. The solid line is the PVDOS-scaled IR spectrum of the OH group (IR spectrum computed using GFN1-xTB).
The PVDOS matrix is not printed to the text output, but only saved to the engine
binary output (.rkf) in the variable Vibrations%PVDOS.
PES point character¶
A PES point can according to the slope and curvature of the PES at that point be classified in the following categories:
- A local minimum on the PES with vanishing nuclear gradients and no negative frequencies.
- A transition state with vanishing nuclear gradients and exactly one negative frequency, i.e. a first order saddle point on the PES.
- A higher order saddle point, i.e. a stationary point on the PES with vanishing nuclear gradients but more than one imaginary frequency.
- A non-stationary point on the PES. Here the gradients are non-zero.
This classification can easily be done if both the gradients and the normal
modes have already been calculated. However, calculating the full Hessian needed
for the entire set of normal modes is very expensive and undesirable if one only
wants to know the character of a PES point. The AMS driver can quickly, and
without calculating the full Hessian, characterize a PES point into one of the
above categories. This can be used to confirm the success of e.g. a
transition state search or geometry
optimization. A PES point can be characterized by
requesting PESPointCharacter as a property:
Properties
PESPointCharacter [True | False]
End
PropertiesPESPointCharacterType: Bool Default value: False GUI name: Characterize PES point Description: Determine whether the sampled PES point is a minimum or saddle point. Note that for large systems this does not entail the calculation of the full Hessian and can therefore be used to quickly confirm the success of a geometry optimization or transition state search.
This will calculate the few lowest normal modes using an iterative diagonalization of the Hessian [1] based on a Davidson algorithm implemented in the PRIMME library [2]. The procedure has been optimized for finding a small number of low-lying eigenvalues in as few matrix-vector multiplications (and thus single point calculations) as possible. This is facilitated by performing the iterative method using a pre-conditioning matrix based on an approximation of the Hessian. The approximate Hessian is obtained from the full Hessian at a lower level of theory. This calculation also provides the initial guesses for the desired normal modes. What the lower level of theory is depends on the main engine used in the calculation: DFTB with the GFN1-xTB model is used as the lower level of theory for relatively slow engines, e.g. DFT based engines. For semi-empirical engines like DFTB or MOPAC, the lower level of theory is currently UFF. It is currently not possible to change the engine used to obtain the preconditioning Hessian and the approximate modes.
- Note that the iterative calculation of the normal modes is skipped when …
- … the nuclear gradients are so large that the PES point is considered non-stationary. The calculation of the modes is then just not necessary for classifying it.
- … the full normal modes or Hessian have also been requested. The iterative calculation is then not necessary, as all modes are already known.
- … the molecule is very small. (For small systems the iterative calculation of the few lowest normal modes is not faster than the full calculation of all modes, so all modes are calculated instead.)
- The classification as a stationary or non-stationary point uses the gradient convergence criterion from the geometry optimizer as the tolerance, see geometry optimization. This makes sure that the criterion for what is considered converged/stationary is always in sync between the optimizer and the PES point characterization.
- For periodic systems the PES point characterization does not take the lattice degrees of freedom into account. A PES point where the nuclear gradients are small enough would for example be classified as a stationary point, even if the system is under stress.
Details of the iterative procedure can be configured in the
PESPointCharacter block:
PESPointCharacter
Displacement float
NumberOfModes integer
Tolerance float
End
PESPointCharacterType: Block Description: Options for the characterization of PES points. DisplacementType: Float Default value: 0.04 Description: Controls the size of the displacements used for numerical differentiation: The displaced geometries are calculated by taking the original coordinates and adding the mass-weighted mode times the reduced mass of the mode times the value of this keyword. NumberOfModesType: Integer Default value: 2 Description: The number of (lowest) eigenvalues that should be checked. ToleranceType: Float Default value: 0.02 Description: Convergence tolerance for residual in iterative Davidson diagonalization.
- Note that the residual tolerance that can be achieved is limited by the numerical differentiation that is performed. The default values should apply in most cases, but if convergence becomes a problem one may choose to increase the tolerance or to increase the step size (slightly). Note that the default residual tolerance is lower than for the other mode selective methods. This is because PRIMME uses a different convergence criteria than mode tracking/refinement. The higher value used as a default will therefore not result in decreased levels of accuracy. The method will bail if the number of iterations exceeds the number of normal modes as at this point still achieving convergence becomes unlikely, in part due to the next point.
- In order to avoid producing the known and irrelevant rigid modes, the method searches for normal modes orthogonal to six (or five) rigid modes. Imperfections due to the numerical differentiation may mean that the translational and rotational rigid modes are not exact eigenmodes of the Hessian that is constructed. As a result, some part of the lowest vibrational normal mode may lie in the span of the theoretical rigid modes and therefore be inaccessible to the Davidson method. This places a lower bound on the residual tolerance that can be achieved, which is directly related to the numerical differentiation accuracy. The take-away: do not set the tolerance too low, the default usually suffices.
- Behind the scenes, the method actually computes a few more modes than requested. In the case of multiplicities, eigenvalues may still converge out of order. These additional eigenvalues essentially guarantee that the obtained modes are indeed the lowest ones.
| [1] |
|
| [2] |
|
Elastic tensor¶
The elastic tensor \(c_{\alpha, \beta}\) (Voigt notation) is computed via second order numerical differentiation of the energy \(E\) WRT strain deformations \(\epsilon_\alpha\) and \(\epsilon_\beta\):
where \(V_0\) is the volume of the unit cell (for 2D periodic system \(V_0\) is the area of the unit cell, and for 1D periodic system \(V_0\) is the length of the unit cell).
For each strain deformation \(\epsilon_\alpha \epsilon_\beta\), the atomic positions will be optimized. The elastic tensor can be computed for any periodicity, i.e. 1D, 2D and 3D.
See also
To compute the elastic tensor, request it in the Properties input block of
AMS:
Properties
ElasticTensor [True | False]
End
Note
The elastic tensor should be computed at the fully optimized geometry. One should therefore perform a geometry optimization of all degrees of freedom, including the lattice vectors. It is recommended to use a tight gradient convergence threshold for the geometry optimization (e.g. 1.0E-4). Note that all this can be done in one job by combining the geometry optimization task with the elastic tensor calculation.
The elastic tensor (in Voigt notation) is printed to the output file and stored
in the engine result file in the AMSResults
section (for 3D system, the elastic tensor in Voigt notation is a 6x6 matrix;
for 2D systems is a 3x3 matrix; for 1D systems is just one number).
Options for the numerical differentiation procedure can be specified in the
ElasticTensor input block:
ElasticTensor
MaxGradientForGeoOpt float
StrainStepSize float
End
ElasticTensorType: Block Description: Options for numerical evaluation of the elastic tensor. MaxGradientForGeoOptType: Float Default value: 0.0001 Unit: Hartree/Angstrom GUI name: Maximum nuclear gradient Description: Maximum nuclear gradient for the relaxation of the internal degrees of freedom of strained systems. StrainStepSizeType: Float Default value: 0.001 Description: Step size (relative) of strain deformations used for computing the elastic tensor numerically.
Pressure or non-isotropic external stress can be included in your simulation via the corresponding engine addons.
The elastic tensor calculation supports AMS’ double parallelization, which can perform the calculations for the individual displacements in parallel. This is configured automatically, but can be further tweaked using the keys in the NumericalDifferentiation%Parallel block:
ElasticTensor
Parallel
nCoresPerGroup integer
nGroups integer
nNodesPerGroup integer
End
End
ElasticTensorParallelType: Block Description: The evaluation of the elastic tensor via numerical differentiation is an embarrassingly parallel problem. Double parallelization allows to split the available processor cores into groups working through all the available tasks in parallel, resulting in a better parallel performance. The keys in this block determine how to split the available processor cores into groups working in parallel. nCoresPerGroupType: Integer GUI name: Cores per task Description: Number of cores in each working group. nGroupsType: Integer Description: Total number of processor groups. This is the number of tasks that will be executed in parallel. nNodesPerGroupType: Integer Description: Number of nodes in each group. This option should only be used on homogeneous compute clusters, where all used compute nodes have the same number of processor cores.
Phonons¶
Collective oscillations of atoms around theirs equilibrium positions, giving rise to lattice vibrations, are called phonons. AMS can calculate phonon dispersion curves within standard harmonic theory, implemented with a finite difference method. Within the harmonic approximation we can calculate the partition function and therefore thermodynamic properties, such as the specific heat and the free energy.
See also
Example: Phonons for graphene, Example: Phonons with isotopes, Example: User-defined Brillouin zone for phonon dispersion and diamond lattice optimization and phonons tutorial
The calculation of phonons is enabled in the Properties block.
Properties
Phonons [True | False]
End
Note
Phonon calculations should be performed on optimized geometries, including the lattice vectors. This can be done by either using an already optimized system as input, or by combining the phonon calculation with the geometry optimization task (you should set the GeometryOptimization%OptimizeLattice input option to True).
The details of the phonon calculations are configured in the
NumericalPhonons block:
NumericalPhonons
SuperCell # Non-standard block. See details.
...
End
StepSize float
DoubleSided [True | False]
UseSymmetry [True | False]
Interpolation integer
NDosEnergies integer
AutomaticBZPath [True | False]
BZPath
Path # Non-standard block. See details.
...
End
End
Parallel
nCoresPerGroup integer
nGroups integer
nNodesPerGroup integer
End
End
NumericalPhononsSuperCellType: Non-standard block Description: Used for the phonon run. The super lattice is expressed in the lattice vectors. Most people will find a diagonal matrix easiest to understand.
The most important setting here is the super cell transformation. In principle this should be as large as possible, as the phonon bandstructure converges with the size of the super cell. In practice one may want to start with a 2x2x2 cell and increase the size of the super cell until the phonon band structure converges:
NumericalPhonons
SuperCell
2 0 0
0 2 0
0 0 2
End
End
By default the phonon dispersion curves are computed for the standard path though the Brillouin zone (see https://doi.org/10.1016/j.commatsci.2010.05.010). One can request the a different path using the following keywords (for an example of how to specify a user-defined path see Example: User-defined Brillouin zone for phonon dispersion):
NumericalPhonons
AutomaticBZPath [True | False]
BZPath
Path # Non-standard block. See details.
...
End
End
End
NumericalPhononsAutomaticBZPathType: Bool Default value: True GUI name: Automatic BZ path Description: If True, compute the phonon dispersion curve for the standard path through the Brillouin zone. If False, you must specify your custom path in the [BZPath] block. BZPathType: Block Description: If [NumericalPhonons%AutomaticBZPath] is false, the phonon dispersion curve will be computed for the user-defined path in the [BZPath] block. You should define the vertices of your path in fractional coordinates (with respect to the reciprocal lattice vectors) in the [Path] sub-block. If you want to make a jump in your path (i.e. have a discontinuous path), you need to specify a new [Path] sub-block. PathType: Non-standard block Recurring: True Description: A section of a k space path. This block should contain multiple lines, and in each line you should specify one vertex of the path in fractional coordinates. Optionally, you can add text labels for your vertices at the end of each line.
Other keywords in the NumericalPhonons block modify the details of the numerical differentiation
procedure and the accuracy of the results:
NumericalPhononsStepSizeType: Float Default value: 0.04 Unit: Angstrom Description: Step size to be taken to obtain the force constants (second derivative) from the analytical gradients numerically. DoubleSidedType: Bool Default value: True Description: By default a two-sided (or quadratic) numerical differentiation of the nuclear gradients is used. Using a single-sided (or linear) numerical differentiation is computationally faster but much less accurate. Note: In older versions of the program only the single-sided option was available. UseSymmetryType: Bool Default value: True Description: Whether or not to exploit the symmetry of the system in the phonon calculation. InterpolationType: Integer Default value: 100 Description: Use interpolation to generate smooth phonon plots. NDosEnergiesType: Integer Default value: 1000 Description: Nr. of energies used to calculate the phonon DOS used to integrate thermodynamic properties. For fast compute engines this may become time limiting and smaller values can be tried.
The numerical phonon calculation supports AMS’ double parallelization, which can perform the calculations for the individual displacements in parallel. This is configured automatically, but can be further tweaked using the keys in the NumericalPhonons%Parallel block:
NumericalPhonons
Parallel
nCoresPerGroup integer
nGroups integer
nNodesPerGroup integer
End
End
NumericalPhononsParallelType: Block Description: Computing the phonons via numerical differentiation is an embarrassingly parallel problem. Double parallelization allows to split the available processor cores into groups working through all the available tasks in parallel, resulting in a better parallel performance. The keys in this block determine how to split the available processor cores into groups working in parallel. Keep in mind that the displacements for a phonon calculation are done on a super-cell system, so that every task requires more memory than the central point calculated using the primitive cell. nCoresPerGroupType: Integer Description: Number of cores in each working group. nGroupsType: Integer Description: Total number of processor groups. This is the number of tasks that will be executed in parallel. nNodesPerGroupType: Integer GUI name: Cores per task Description: Number of nodes in each group. This option should only be used on homogeneous compute clusters, where all used compute nodes have the same number of processor cores.
Numerical differentiation options¶
The following options apply whenever AMS computes gradients, Hessians or stress tensors via numerical differentiation.
NumericalDifferentiation
NuclearStepSize float
StrainStepSize float
UseSymmetry [True | False]
End
NumericalDifferentiationType: Block Description: Define options for numerical differentiations, that is the numerical calculation of gradients, Hessian and the stress tensor for periodic systems. NuclearStepSizeType: Float Default value: 0.005 Unit: Bohr Description: Step size for numerical nuclear gradient calculation. StrainStepSizeType: Float Default value: 0.001 Description: Step size (relative) for numerical stress tensor calculation. UseSymmetryType: Bool Default value: True Description: Whether or not to exploit the symmetry of the system for numerical differentiations.
AMS may use symmetry (key NumericalDifferentiation%UseSymmetry) in case of numerical differentiation calculations.
If symmetry is used only symmetry unique atoms are displaced.
Symmetry is only recognized if the starting geometry has symmetry.
Symmetry is only used for molecules if the molecule has a specific orientation in space, like that the z-axis is the main rotation axis.
If the GUI is used one can click the Symmetrize button (the star), such that the GUI will (try to) symmetrize and reorient the molecule.
There are some cases that even after such symmetrization, the orientation of the molecule is not what is needed for the symmetry to be used in case of numerical differentiation calculations. If that is the case or if key NumericalDifferentiation%UseSymmetry is set to ‘False’, then no symmetry will be used.
The numerical differentiation calculation supports AMS’ double parallelization, which can perform the calculations for the individual displacements in parallel. This is configured automatically, but can be further tweaked using the keys in the NumericalDifferentiation%Parallel block:
NumericalDifferentiation
Parallel
nCoresPerGroup integer
nGroups integer
nNodesPerGroup integer
End
End
NumericalDifferentiationParallelType: Block Description: Numerical differentiation is an embarrassingly parallel problem. Double parallelization allows to split the available processor cores into groups working through all the available tasks in parallel, resulting in a better parallel performance. The keys in this block determine how to split the available processor cores into groups working in parallel. nCoresPerGroupType: Integer Description: Number of cores in each working group. nGroupsType: Integer Description: Total number of processor groups. This is the number of tasks that will be executed in parallel. nNodesPerGroupType: Integer Description: Number of nodes in each group. This option should only be used on homogeneous compute clusters, where all used compute nodes have the same number of processor cores.
Other Properties¶
Properties
DipoleMoment [True | False]
BondOrders [True | False]
End
PropertiesDipoleMomentType: Bool Default value: False Description: Requests the engine to calculate the dipole moment of the molecule. This can only be requested for non-periodic systems. BondOrdersType: Bool Default value: False Description: Requests the engine to calculate bond orders. For MM engines these might just be the defined bond orders that go into the force-field, while for QM engines, this might trigger a bond order analysis based on the electronic structure.