Comparison of systems¶
Simple methods for comparing two ChemicalSystem instances:
- ChemicalSystem.has_same_atoms(other: ChemicalSystem, attributes: List[Literal['gui', 'adf', 'band', 'forcefield', 'dftb', 'reaxff', 'qe', 'vasp']] = []) bool
- ChemicalSystem.has_same_atoms(other: ChemicalSystem, comp: atom_comparator_func_t) bool
Checks if two systems have identical atoms in the same order.
Atoms are compared one by one using either a user-defined comparator function, or the
Atom.has_identical_attributesmethod with a user defined list of (optional) attributes groups to consider in the comparison.
- ChemicalSystem.has_same_coords(other: ChemicalSystem, tol: float = 0.001, unit: str = 'angstrom') bool
Checks if the atomic coordinates of two systems are within a threshold of each other.
This check is intended for systems that have the same number of atoms and all atoms in the same order. (You likely want to call
has_same_atomsbefore calling this method.) The thresholdtolis compared against the distance between two corresponding atoms. This ensures that the return value of this method does not depend on an overall rotation of the two systems.
- ChemicalSystem.has_same_geometry(other: ChemicalSystem, tol: float = 0.001, unit: str = 'angstrom') bool
Checks if the atomic coordinates and lattice vectors of two systems are within a threshold of each other. This is just a shorthand for calling
has_same_coordsandlattice.is_close()on the two systems.
- ChemicalSystem.has_same_regions(other: ChemicalSystem) bool
Checks if two systems have identical regions, meaning region names match and each region includes the same atoms.
This only checks the indices of the atoms assigned to the different regions. It does not check if atoms with the same index are actually the same. Use
has_same_atomsfor that.
- ChemicalSystem.has_same_selection(other: ChemicalSystem, consider_selection_order: bool = False) bool
Checks if two systems have the same atom selection.
This only checks the indices of the selected atoms. It does not check if atoms with the same index are actually the same. Use
has_same_atomsfor that.By default the selection order is ignored in the check and the two selections are compared as a set. This can be changed via the
consider_selection_orderargument.
Atomic and Molecular hashing (fingerprints)¶
The ChemicalSystem provides molecular hashing (fingerprinting) capabilities that generate unique integer identifiers for molecules and individual atoms based on their chemical structure and environment. These hashes enable molecule comparison, detection of chemically equivalent atoms, and comparison of atomic chemical environments.
The hashes are invariant to atom ordering and molecular rotation/translation, but capture the essential chemical topology.
Note
Bonding information is required for hashing to work. Call guess_bonds() first if bonds aren’t defined!
Why use molecular hashing?
Molecule comparison / identification: Efficiently determine if two molecules are identical or different. The same molecule produces the same hash regardless of atom ordering or atomic coordinates (unless stereochemistry options are enabled, see below).
Chemical equivalence detection: Identify atoms in symmetric or equivalent chemical environments
Basic Examples¶
Example 1: Hash invariance to atom ordering and geometry
The molecular hash is the same regardless of how atoms are ordered or how the molecule is positioned in space. This can be useful if you want to check if two molecules with different geometry correspond to the “same system”, even if you cannot assume that the atoms are in the same order.
from scm.base import ChemicalSystem
import random
# Create a simple molecule: ethanol
mol_a = ChemicalSystem.from_smiles("OCC")
mol_a.guess_bonds()
print(f"{'Hash of original ethanol':<40}: {mol_a.hash()}")
# Let's make a copy and modify it a bit
mol_b = mol_a.copy()
shuffle_indices = list(range(mol_b.num_atoms))
random.shuffle(shuffle_indices)
mol_b.reorder_atoms(shuffle_indices)
mol_b.perturb_coordinates(0.1) # randomly perturb coordinates
print(f"{'Hash of shuffled and perturbed ethanol':<40}: {mol_b.hash()}")
# But remove an atom or break a bond, and the hash changes:
mol_c = mol_a.copy()
mol_c.bonds.remove_bond(0)
print(f"{'Hash ethanol with a bond removed':<40}: {mol_c.hash()}")
Example 2: Using atomic hashes to identify chemical environments
Atomic hashes reveal which atoms are in equivalent chemical environments. The depth parameter
controls how much of the surrounding structure is considered. The various optional parameters determine
which features will be considered.
from scm.base import ChemicalSystem
# Molecule with two methyl groups and various hydrogens
mol = ChemicalSystem(
"""
System
Atoms
C -4.0419 0.5780 -0.4490 # 0 Methyl group 1
H -3.4703 0.0193 -1.2206 # 1
H -5.0855 0.7047 -0.8071 # 2
H -4.0585 -0.0070 0.4950 # 3
C -1.1373 3.1240 0.2318 # 4 Methyl group 2
H -0.1113 2.9084 0.5999 # 5
H -1.6136 3.8633 0.9089 # 6
H -1.0664 3.5552 -0.7887 # 7
C -3.4113 1.9507 -0.2140 # 8 Middle C
H -3.9782 2.4815 0.5805 # 9
H -3.4937 2.5415 -1.1513 # 10
C -1.9597 1.8726 0.1904 # 11 C connected to O
O -1.4448 0.8029 0.4844 # 12
End
End
"""
)
mol.guess_bonds()
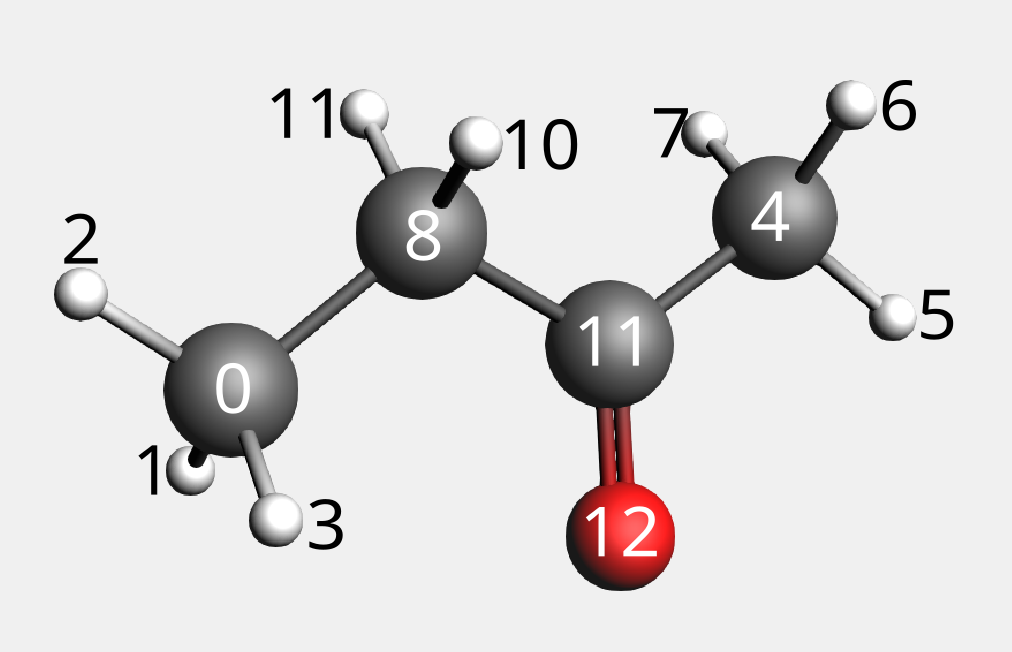
In the following picture we show the indices of the atoms.
Now let’s examine how the atomic hashes change using different depths;
# ==== Full molecular context (depth=-1, the default) ====
hashes_full = mol.atomic_hashes(depth=-1)
# Hydrogens within the same methyl group are equivalent
assert hashes_full[1] == hashes_full[2] == hashes_full[3] # Methyl 1 H's
assert hashes_full[5] == hashes_full[6] == hashes_full[7] # Methyl 2 H's
# But hydrogens from different methyl groups are NOT equivalent (different molecular position)
assert hashes_full[1] != hashes_full[5]
# The two methyl carbons are also different
assert hashes_full[0] != hashes_full[4]
# ==== Now with depth=0 (only local properties) ====
hashes_local = mol.atomic_hashes(depth=0)
# Both methyl carbons now look the same (both are C with 4 bonds)
assert hashes_local[0] == hashes_local[4] == hashes_local[8]
# All methyl hydrogens are now equivalent (all bonded to C)
for h_idx in [1, 2, 3, 5, 6, 7, 9, 10]:
assert hashes_local[1] == hashes_local[h_idx]
# But the carbon connected to O is still different (different neighbors)
assert hashes_local[0] != hashes_local[11]
# ==== With depth=2, we see intermediate behavior ====
hashes_d2 = mol.atomic_hashes(depth=2)
# Methyl 1 hydrogens are still equivalent
assert hashes_d2[1] == hashes_d2[2] == hashes_d2[3]
# But now different from methyl 2 (within 2 bonds they "see" different environments)
assert hashes_d2[1] != hashes_d2[5]
Example 3: Effect of hashing options
Different options capture different aspects of molecular structure.
Most of the options relate to connectivity (bonds), but rs_stereo and ez_stereo look at the 3D information to distinguish between isomers or enantiomers.
from scm.base import ChemicalSystem
# Lactic acid enantiomers
d_lactic = ChemicalSystem.from_smiles("C[C@H](C(=O)O)O")
l_lactic = ChemicalSystem.from_smiles("C[C@@H](C(=O)O)O")
print("With rs_stereo=False, the enantiomers are identical:")
print(f" d_lactic hash : {d_lactic.hash(rs_stereo=False)}")
print(f" l_lactic hash : {l_lactic.hash(rs_stereo=False)}")
print("With rs_stereo=True, the enantiomers are different:")
print(f" d_lactic hash : {d_lactic.hash(rs_stereo=True)}")
print(f" l_lactic hash : {l_lactic.hash(rs_stereo=True)}")
# 2-Butene cis/trans isomers:
cis = ChemicalSystem.from_smiles("C/C=C\C")
trans = ChemicalSystem.from_smiles("C/C=C/C")
print("With ez_stereo=False, the isomers are identical:")
print(f" cis hash : {cis.hash(ez_stereo=False)}")
print(f" trans hash : {trans.hash(ez_stereo=False)}")
print("With ez_stereo=True, the isomers are different:")
print(f" cis hash : {cis.hash(ez_stereo=True)}")
print(f" trans hash : {trans.hash(ez_stereo=True)}")
How it works¶
The hashing algorithm operates in three stages:
Local atomic hashing: Each atom receives an initial hash based on its element type and local chemical properties (coordination number, bond orders, ring membership, etc.)
Stereochemistry incorporation (optional): The hashes are updated to include stereochemical information for double bonds (E/Z isomerism) and chiral centers (R/S configuration)
Environment propagation (optional, enabled by default): Each atom’s hash is combined with hashes from neighboring atoms at increasing distances, allowing the hash to capture increasingly larger regions of the molecular graph
The molecular hash is computed by summing all atomic hashes, producing a single integer that characterizes the entire structure.
Important notes:
Hash values are deterministic but arbitrary — the numerical value itself has no chemical meaning, and small structural changes produce completely different hash values
Hashes are not smooth features suitable for machine learning; they are designed for exact structure matching
The algorithm requires bonding information — use
guess_bonds()before hashing if bonds aren’t already definedHash collisions are theoretically possible but extremely rare with appropriate options
Conformers will all have the same hash values. Enantiomers / Isomers will have different values if
rs_stereoandez_stereoare enabled
Recommended settings:
Basic molecular comparison: Use defaults
Stereochemistry-aware comparison: Enable
ez_stereo=Trueandrs_stereo=TrueDetailed chemical environment: Enable
bond_orders=Trueandring=TrueFor comparing local chemical environment of atoms: Use a finte
depth, typically 1 or 2 (but depends on your application)
Note
This hashing functionality replaces the molsg library from earlier versions of the Amsterdam Modeling Suite. The new implementation is integrated directly into the ChemicalSystem class, eliminating the need for separate molsg calculations.
Despite the hashing algorithm following the same general principles, the actual hash values between the ChemicalSystem and the molsg library will be different.
API Reference¶
- ChemicalSystem.hash(depth: int = -1, coordination: bool = True, bond_orders: bool = False, ez_stereo: bool = False, rs_stereo: bool = False, ring: bool = False, implicit_h: bool = False) int
Compute a hash-based identifier for the entire molecule.
Generates a single integer hash value that uniquely identifies the molecular structure. This hash is computed by combining the atomic hashes (see
atomic_hashes()) of all atoms in the molecule. Two molecules with identical structures/connectivity will produce the same hash value regardless of atom ordering, coordinates (rotation/translation).The hashing algorithm is based on molecular connectivity (bonding topology). By default, different conformations of the same molecule (same bonds, different 3D coordinates) produce the same hash. However, when stereochemistry options (
ez_stereo,rs_stereo) are enabled, stereoisomers (cis/trans, R/S enantiomers) are distinguished and produce different hash values.- Parameters:
depth: Hash propagation depth through the molecular graph (controls how much structural information is incorporated):-1 (default): Unlimited propagation - the hash reflects the complete molecular structure. Recommended for whole-molecule comparison.
0: No propagation - hash based only on local atomic properties without considering connectivity patterns beyond immediate neighbors.
n > 0: Propagate information up to n bonds away. Smaller values produce hashes that are less sensitive to distant structural features.
coordination: Include coordination number (number of bonds per atom) in the hash. Enabled by default. Essential for distinguishing most structural differences.bond_orders: Include bond order information (single/double/triple bonds). When disabled (default), molecules differing only in bond orders may produce the same hash. Enable to distinguish resonance structures or different bond order assignments.ez_stereo: Include E/Z stereochemistry around double bonds. When enabled, cis and trans isomers produce different hashes. Requires meaningful 3D coordinates to determine stereochemical configuration.rs_stereo: Include R/S stereochemistry at chiral centers. When enabled, enantiomers produce different hashes. Requires meaningful 3D coordinates to determine absolute configuration.ring: Include ring membership information. When enabled, distinguishes atoms in rings from acyclic atoms and incorporates ring sizes.implicit_h: Treat hydrogen atoms implicitly. When enabled, hydrogen atoms contribute zero to the hash, and non-hydrogen atoms include a contribution based on bonded hydrogens. Useful for comparing structures with different hydrogen representations (explicit vs. implicit).
- Returns:
Integer hash value identifying the molecular structure. The hash is deterministic and reproducible but the numerical value itself is arbitrary.
- Notes:
Requires bonding information: Call
guess_bonds()first if bonds aren’t definedAtom-ordering invariant: Reordering atoms in the molecule produces the same hash
Coordinate invariant (by default): Rotating, translating, or reflecting the molecule produces the same hash (unless
ez_stereoorrs_stereoare enabled)Parameter consistency critical: When comparing molecules, use identical parameter settings for both. Different parameters will produce incomparable hash values.
Collisions extremely rare: With default settings, hash collisions between different molecules are theoretically possible but exceptionally unlikely for typical organic molecules
- Warnings:
Comparing hashes computed with different parameter settings is meaningless
Stereochemistry options require valid 3D coordinates; incorrect coordinates may produce unreliable stereochemical assignments
- See Also:
atomic_hashes(): Compute individual hashes for each atom
- ChemicalSystem.atomic_hashes(depth: int = -1, coordination: bool = True, bond_orders: bool = False, ez_stereo: bool = False, rs_stereo: bool = False, ring: bool = False, implicit_h: bool = False) List[int]
Compute hash-based atomic structure identifiers for all atoms.
Generates unique integer hashes for each atom based on their local chemical environment. These hashes enable identification of chemically equivalent atoms, substructure matching, and atom-level structure comparison. The hashes are designed for exact structure identification and isomorphism detection - NOT for machine learning features (the hash values are not “smooth” and small structural changes produce completely different values).
The hashing algorithm combines atomic properties (element type, coordination, bond orders, etc.) with information from the surrounding molecular graph. By default, each atom’s hash reflects its complete molecular environment.
- Parameters:
depth: Hash propagation depth through the molecular graph (controls how much of the molecular environment is incorporated into each atom’s hash):-1 (default): Unlimited propagation - each atom’s hash incorporates information from the entire molecule, making hashes sensitive to the complete molecular structure.
0: No propagation - hashes reflect only immediate atomic properties (element, coordination number, bond orders, etc.) without considering neighbors.
n > 0: Propagate information up to n bonds away - hashes incorporate environment information from atoms within n bonds, enabling local similarity comparisons while ignoring distant parts of the molecule.
coordination: Include coordination number (number of bonds) in the hash. Enabled by default. Distinguishes atoms with different bonding patterns.bond_orders: Include bond order information (single/double/triple bonds). When enabled, the hash distinguishes between C-C, C=C, and C≡C bonding. Bond orders are mapped to standard values (1.0, 1.5, 2.0, 3.0, etc.).ez_stereo: Include E/Z stereochemistry for double bonds. When enabled, cis and trans isomers produce different hashes. Requires 3D coordinates to determine stereochemical configuration.rs_stereo: Include R/S stereochemistry for chiral centers. When enabled, enantiomers produce different hashes. Requires 3D coordinates to determine handedness at tetrahedral stereocenters.ring: Include ring membership information. When enabled, atoms in rings are distinguished from non-ring atoms, and the smallest ring size is incorporated into the hash.implicit_h: Treat hydrogen atoms implicitly. When enabled:All hydrogen atoms receive a hash value of zero
Non-hydrogen atoms get a contribution based on the number of bonded hydrogens
Useful for comparing structures with different hydrogen representations
- Returns:
List of integers (one per atom). Atoms in identical chemical environments have identical hash values. Hash values are deterministic and reproducible but arbitrary as numbers (a small structural difference typically produces a completely different hash value).
- Notes:
Requires bonding information: Call
guess_bonds()first if bonds aren’t definedHash ordering invariance: Reordering atoms produces the same hash values (though in different positions in the returned list)
Parameter consistency: When comparing atoms across molecules, use identical parameter settings
Stereochemistry requires 3D: The
ez_stereoandrs_stereooptions require meaningful 3D coordinatesNot for ML: These are discrete identifiers, not continuous features suitable for machine learning
- See Also:
hash(): Compute a single hash for the entire molecule