Using active atoms¶
The active_atoms keyword argument can be very useful for many types of reaction mapping problems. active_atoms is a user-supplied list of atom indices that indicate which atoms are allowed to be mapped to an atom with another index. The complement of this set, the inactive atoms, are those atoms that must be mapped to the same index (e.g., inactive reactant atom 5 must be mapped to inactive product atom 5). Active atoms can be useful to add constraints to the problem or to reduce the complexity of a problem for which there is only a small reactive region. For this example, we’ll take a look at the following Claisen rearrangement mapping with active atoms:
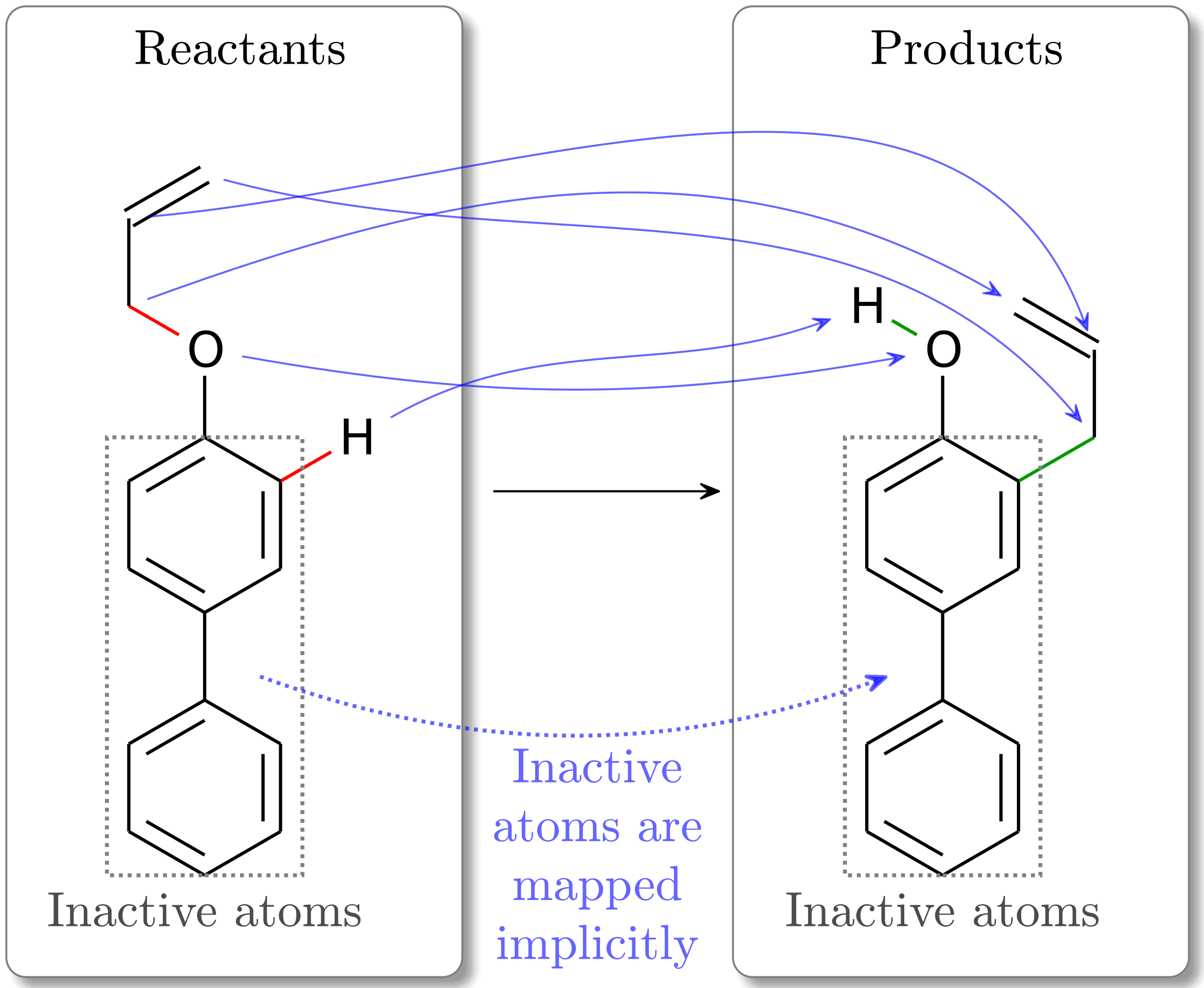
Fig. 2 In this Claisen rearrangement mapping, we see the inactive atoms (in the gray dashed boxes) in the reactants are mapped directly to the identical index in the products. The active atoms, however, can be mapped to a differently indexed product atom, which is observed to happen in this example.¶
Below we also provide the Python code for this example. The input is given using the atoms and bonds format, which is often preferable when using active atoms.
Note
It is often easier to use the atoms and bonds inputs for a reactmap.Molecule when using active_atoms. This is because active_atoms requires input of atom indices, and it’s easiest to keep track of these when using list input for atoms and bonds.
import scm.reactmap as rm
reactant = rm.Molecule(atoms=['C','H','H','C','H','C','H','H','O','C','C','H','C','H','C','C','H','C','H','C','C','H','C','H','C','H','C','H','C','H'], bonds=[(0,1),(0,2),(0,3),(3,4),(3,5),(5,6),(5,7),(5,8),(8,9),(9,10),(10,11),(10,12),(12,13),(12,14),(14,15),(15,16),(15,17),(17,18),(17,9),(14,19),(19,20),(20,21),(20,22),(22,23),(22,24),(24,25),(24,26),(26,27),(26,28),(28,29),(28,19)],active_atoms=[0,1,2,3,4,5,6,7,8,18])
product = rm.Molecule(atoms=['O','C','H','H','C','H','C','H','H','C','C','H','C','H','C','C','H','C','H','C','C','H','C','H','C','H','C','H','C','H'],bonds=[(18,0),(0,9),(1,17),(1,2),(1,3),(1,4),(4,5),(4,6),(6,7),(6,8),(9,10),(10,11),(10,12),(12,13),(12,14),(14,15),(15,16),(15,17),(17,9),(14,19),(19,20),(20,21),(20,22),(22,23),(22,24),(24,25),(24,26),(26,27),(26,28),(28,29),(28,19)],active_atoms=[0,1,2,3,4,5,6,7,8,18])
reaction = rm.Reaction(reactant=reactant, product=product, name="claisen rearrangement")
rm.Map(reaction)
print("Mapping cost:", reaction.cost)
print("active atom map")
for idx in reactant.active_atoms:
print("Reactant atom {:2} -> Product atom {:2}".format(idx,reaction.mapping[idx]))
This produces the following output:
Mapping cost: 4
active atom map
Reactant atom 0 -> Product atom 1
Reactant atom 1 -> Product atom 3
Reactant atom 2 -> Product atom 2
Reactant atom 3 -> Product atom 4
Reactant atom 4 -> Product atom 5
Reactant atom 5 -> Product atom 6
Reactant atom 6 -> Product atom 7
Reactant atom 7 -> Product atom 8
Reactant atom 8 -> Product atom 0
Reactant atom 18 -> Product atom 18