Using active bonds¶
The active_bonds keyword argument allows you to add another type of constraint to reaction mapping problems. Specifically, the active_bonds indicate which bonds are allowed to be broken/formed during a reaction. It is often useful to think of this set in terms of its complement, the inactive bonds, which are those bonds that must be preserved in the reactant and must not be created in the product. Active bonds can be used to help reduce a problem’s complexity or to add a chemically meaningful constraint (e.g., no spontaneous breaking of a C-C bond in a reaction mechanism).
For this example, we will investigate a Favorskii rearrangement reaction. Solving this reaction mapping problem with no active_bonds information (i.e., no additional constraints), we obtain the following mapping:
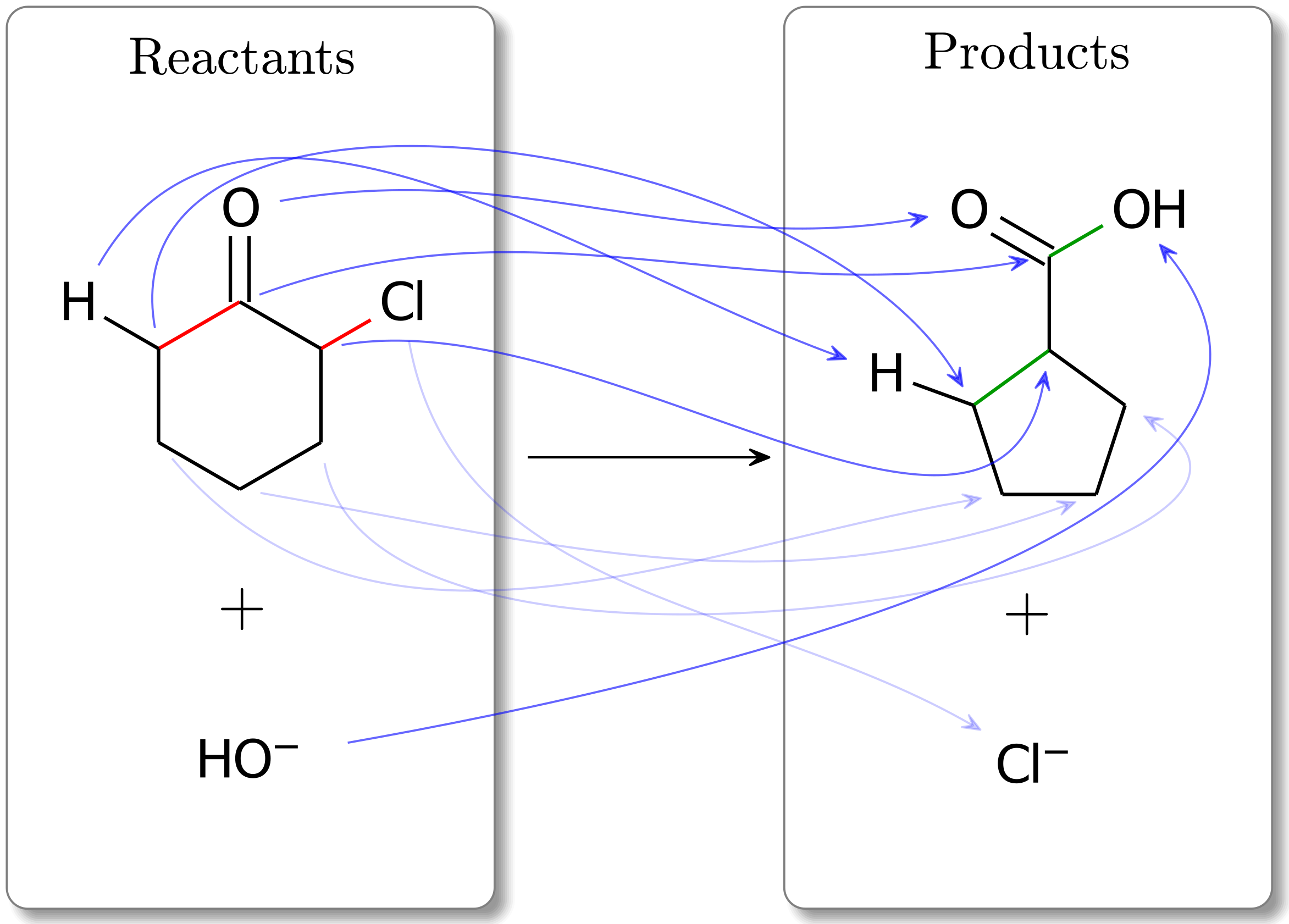
Fig. 3 The optimal atom mapping (cost: 4) for a Favorskii rearrangement for an unconstrained problem (i.e., no active bonds information is used). Note that some of the mapping arrows represent C and neighboring H atoms. Additionally, some arrows are made more transparent to reduce complexity.¶
Though this atom mapping is the optimal solution, the implied mechanism is not consistent with our chemical intuition. Specifically, we do not expect the non-nucleophilic -CH2- group to attack another carbon, causing the Cl atom to leave. It is more likely that this reaction proceeds first with a deprotonation of one of the H atoms in the -CH2- group, and then a successive attack of the carbon atom. One way to ensure we observe a deprotonation is to make the CH2- carbonyl bond inactive. Doing so yields a reaction mapping with a higher cost but one that is more consistent with our expectations. This is shown below.
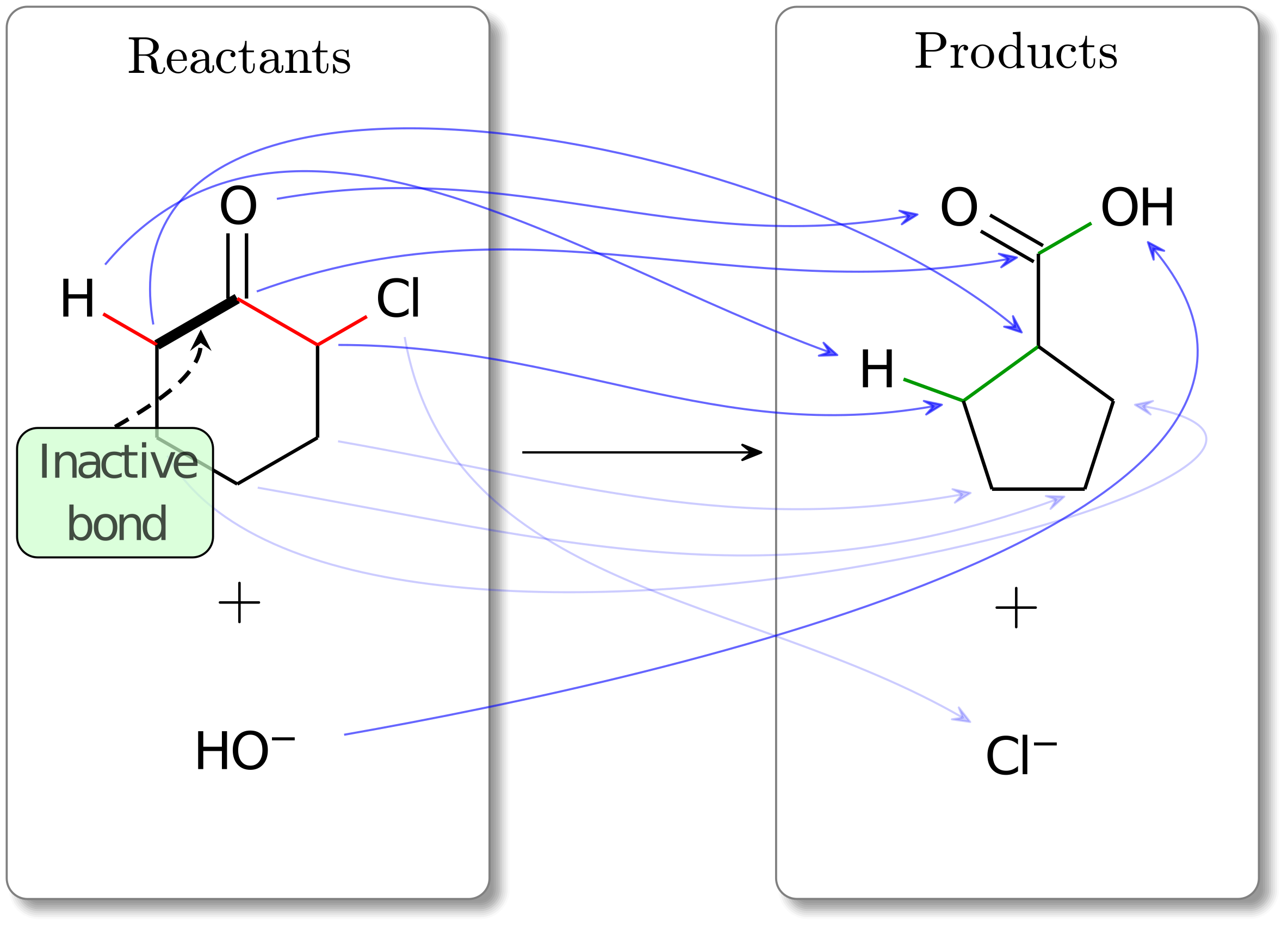
Fig. 4 The optimal atom mapping (cost: 6) for a Favorskii rearrangement for a constrained problem (i.e., active bonds information is used). The bond in bold is treated as an inactive bond in this problem, meaning it cannot be broken. Preserving this bond leads to an atom mapping with a deprotonation step in the implied mechanism, which is consistent with the true mechanism of this reaction.¶
Below we also provide the Python code for this example. The input is given using the atoms and bonds format, which is often preferable when using active bonds.
Note
It is often easier to use the atoms and bonds inputs for a reactmap.Molecule when using active_bonds. This is because active_bonds requires input of atom indices, and it’s easiest to keep track of these when using list input for atoms and bonds.
import scm.reactmap as rm
settings = rm.Settings(use_heuristics = False)
atoms_R = ['C','H','H','C','H','H','C','H','H','C','H','H','C','H','Cl','C','O','O','H']
bonds_R = [(0,1),(0,2),(0,3),(3,4),(3,5),(3,6),(6,7),(6,8),(6,9),(9,10),(9,11),(9,12),(12,13),(12,14),(12,15),(15,16),(0,15),(17,18)]
atoms_P = ['C','H','H','C','H','H','C','H','H','C','H','H','C','H','C','O','O','H','Cl']
bonds_P = [(0,1),(0,2),(0,3),(3,4),(3,5),(3,6),(6,7),(6,8),(6,9),(9,10),(9,11),(9,12),(12,0),(12,13),(12,14),(14,15),(14,16),(16,17)]
active_R = list( set(bonds_R) - set([(0,15)]) )
reactant = rm.Molecule(atoms=atoms_R, bonds=bonds_R )
product = rm.Molecule(atoms=atoms_P, bonds=bonds_P)
reaction = rm.Reaction(reactant=reactant, product=product, name="Favorskii")
rm.Map(reaction)
print("Mapping cost (unconstrained):", reaction.cost)
reactant = rm.Molecule(atoms=atoms_R, bonds=bonds_R , active_bonds=active_R)
product = rm.Molecule(atoms=atoms_P, bonds=bonds_P)
reaction = rm.Reaction(reactant=reactant, product=product, name="Favorskii")
rm.Map(reaction)
print("Mapping cost (constrained) :", reaction.cost)
This produces the following output:
Mapping cost (unconstrained): 4
Mapping cost (constrained) : 6