Input: The Essentials¶
Overview¶
Compared to a standard ADF run, there are two additional input components required to run an ADF QM/MM simulation:
- The QMMM key block has to occur in the ADF input file
- A separate force field parameter file.
In this section we describe how to set up an ADF QM/MM simulation, assuming an appropriate force field file is already available. A full description of all of the options and of the FORCE FIELD file is provided in later sections.
Example Input¶
Let Figure 2-1 show the molecular system that we want to simulate and the intended partitioning. Note that this is the same system as shown in Figure 1-1 except that a non-bonded water molecule has been added to the MM region.
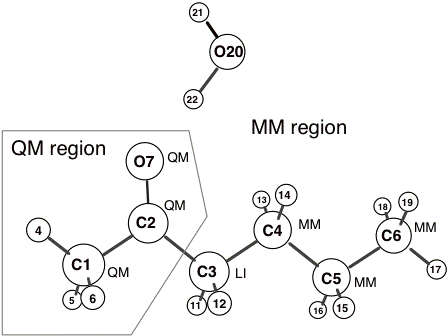
Figure 2-1. Sample structure. Atom numberings and the QM/MM atom types (QM, MM and LI) are shown.
Example 2.1 below shows the ADF input file for the simulation with the geometry defined in Cartesian coordinates. The initial atomic coordinates of the full QM/MM system are defined as they would be in a normal ADF run with the ATOMS and GEOVAR key blocks. Notice that the subkey blocks within the QMMM key block (e.g. MM_CONNECTION_TABLE)****end with SUBEND. If they were closed by ‘END’ ADF would consider the QMMM key block complete at that point.
Example 2.1:
Title example QM/MM input
Fragments
C C.t21
H H.t21
O O.t21
End
Symmetry NOSYM
Charge 0 0
ATOMS
1 C 0.00000 0.00000 0.00000
2 O 1.48700 0.00000 0.00000
3 C -0.76430 1.32381 0.00000
4 C -0.76428 -1.32378 -0.00002
5 H -0.50028 -1.89680 -0.89230
6 H -0.50028 -1.89683 0.89224
7 H -1.83868 -1.12409 -0.00002
8 H -1.40364 1.39261 -0.88358
9 H -1.40364 1.39261 0.88358
10 C 0.22461 2.48208 0.00000
11 H 0.85235 2.42365 -0.89260
12 H 0.85235 2.42365 0.89260
13 C -0.53689 3.80103 0.00000
14 H -1.16478 3.85787 -0.89260
15 H -1.16478 3.85787 0.89260
16 C 0.45222 4.95913 -0.00000
17 H 1.08538 4.90988 -0.88932
18 H 1.08538 4.90988 0.88932
19 H -0.08590 5.91026 0.00000
20 H 2.43700 1.64545 0.00000
21 O 3.03926 2.50556 -0.00000
22 H 3.96191 2.28678 -0.45094
END
QMMM
FORCE_FIELD_FILE sybyl.ff
RESTART_FILE mm.restart
OUTPUT_LEVEL=1
WARNING_LEVEL=1
ELSTAT_COUPLING_MODEL=0
LINK_BONDS
1 - 3 1.38000 H
SUBEND
MM_CONNECTION_TABLE
1 C_2 QM 2 3 4
2 O_2 QM 1
3 C_3 LI 1 8 9 10
4 C_3 QM 1 5 6 7
5 H QM 4
6 H QM 4
7 H QM 4
8 H MM 3
9 H MM 3
10 C_3 MM 3 11 12 13
11 H MM 10
12 H MM 10
13 C_3 MM 10 14 15 16
14 H MM 13
15 H MM 13
16 C_3 MM 13 17 18 19
17 H MM 16
18 H MM 16
19 H MM 16
20 H MM 21
21 O_3 MM 20 22
22 H MM 21
SUBEND
END
GEOMETRY
ITERATIONS 20
CONVERGE E=1.0E-3 GRAD=0.0005
STEP RAD=0.3 ANGLE=5.0
DIIS N=5 OK=0.1 CYC=3
END
XC
LDA VWN
GGA POSTSCF Becke Perdew
End
Integration 3.0
SCF
Iterations 60
Converge 1.0E-06 1.0E-6
Mixing 0.20
End
End Input
Defining the Coordinates¶
The initial coordinates of the full QM/MM system are defined with the ATOMS and GEOVAR key blocks as in a normal ADF run. All input methods allowed by ADF can be used with the exception of the new MOPAC option introduced in ADF1999. For example the coordinates can be defined in Cartesian coordinates as in the example above, by a Z-matrix or by the mixed Z-CART method. It is important to realize that only the initial coordinates of the full or real system are required as input. There is no need to define the coordinates of the QM model system or of the dummy capping atoms. The partitioning of the system into QM and MM regions, and the parameters required to define the capping atoms are given in another part of the input. The program will automatically generate the QM model system and the position of the capping atoms.
IMPORTANT: There is a strict rule concerning the order of the atoms in the ADF input during a QM/MM simulation. Namely, the QM atoms and the LI atoms must precede any MM atoms in the input. The QM and LI atoms can come in any order as long as they come before any MM atoms. The program will check this and abort if this is violated.
ADF atoms and fragments
The typical ATOMS key block in ADF has the following format:
...
ATOMS
{n} atom coordinates {f=fragment}
END
For the QM atoms, the atom labels and fragments should be defined as in a normal ADF run. The atom labels for the MM atoms are not read by the program. Instead, the MM force field atom types for all atoms are defined in the MM_CONNECTION_TABLE subkey block. Similarly if fragments are defined for the MM atoms, the program also ignores these. This is also true of the LI atoms, since in the calculation of the QM model system, capping atoms replace link atoms. The replacement atom type used for the electronic structure calculation is defined in the LINK_BONDS subkey block.
QMMM key block¶
The QMMM key block, which is in bold face in example 2.1, is a mandatory key block. This key block is necessary to invoke a combined QM/MM simulation. It contains the connection table and the force field atom types needed to define the molecular mechanics potential. If link bonds are present then it defines the necessary parameters for each link bond.
This section also explains how to specify the Force Field file to use and various other options.
FORCE_FIELD_FILE- Keyword (required, default = amber95.ff) This keyword simply defines the full path of the force field file to be used for the molecular mechanics potential. The location of the force field file is given after the keyword. The full path can be given, or just the file name. In the latter case, the program looks in the current directory that ADF is executing in. Examples:
FORCE_FIELD_FILE /home/username/sybyl.ff
FORCE_FIELD_FILE sybyl.ff
MM_CONNECTION_TABLESubkey block (required) This key block defines the connection table, the force field atom types and the partitioning of the full system into QM and MM regions. It is critical that the atoms specified in this key block are in the same order as in the ATOMS key block. This is important, because the program may not detect this type of input error and you would get ridiculous results.
MM_CONNECTION_TABLE n FF_LABEL MM_TYPE connection numbers ... END
The labels are defined in the following table
Input column 1 N atom number 2 FF_LABEL Force field atom type. These labels correspond to the atom types defined in the force field file. They can be up to four characters long. Xx defines dummy atoms. 3 MM_TYPE QM, MM or LI 4- connection These define to which atoms the current atom has a covalent bond. numbers These connections are used to generate the molecular mechanics potential. Currently, a maximum of 6 connections is allowed per atom. The connection table should be a fully redundant one. In other words, if atom #1 is bonded to atom #5, they each should have the other atom listed in their connections.
Example:
1 C_2 QM 2 3 4 5 2 O_2 QM 1 3 H QM 1 4 C_3 QM 1 5 6 7 5 Cu QM 4 1 6 H QM 4
A fully non-redundant connection table is also supported. In such a connection table, once a bond is mentioned, it is not mentioned again. In other words, the connection list for any atom cannot contain an atom, which precedes it in the atom numbering.
Example:
1 C_2 QM 2 3 4 5 2 O_2 QM 3 H QM 4 C_3 QM 5 6 7 5 Cu QM 6 H QM
These two connection tables are equivalent. Connection tables that are semi-redundant might cause problems. We recommend the fully redundant connection table.
LINK_BONDSRequired for systems with LINK bonds This key block required for systems with covalent bonds that cross the QM/MM boundary. These bonds are referred to in this document as the link-bonds. Each link bond has a constant parameter ‘alpha‘ associated with it, which is defined as the ratio of the bond length in the real system and of the capping bond in the model QM system. See Section 1 or reference [1] more details. To determine the alpha parameters for each link bond, one can take the capping atom bond distance in a ‘pure QM’ calculation of the QM model system and ratio it to the corresponding bond distance in the real system. These ratios are typically around 1.30 to 1.50 when hydrogen as a capping atom. The LINK_BONDS subkey block has the following format:
LINK_BONDS atom_a - atom_b alpha replacement_fragment [addremove_force_field_type] ... SUBEND
Example:
LINK_BONDS 15 - 3 1.42 H H1 8 - 1 1.40 Cl.dzp Cl SUBEND
The integers atom_a and atom_b refer to the numbering of the two atoms involved in the link bond. One of the atoms will be a LI type atom whereas the other will be a QM type atom. Atom_a and atom_b must be separated by ” - ” with at least one space between the integer and the hyphen. In other words ‘3 - 4’ is OK, but not ‘3- 4’ ‘3 -4’. Atoms need not be in any particular order, and the order of the link bonds is also not important. Following this is the alpha parameter for that specific bond. The replacement_fragment is the ADF atom used for the capping atom in the electronic structure calculation of the QM model system. Often the capping atom is a hydrogen atom, however, it need not be. The replacement_fragment must be present in the FRAGMENT key block in the ADF input file. The addremove_force_field_type need only be present for the AddRemove model [3], and indicates the force field type of the capping atom (similar to FF_LABEL in the MM_CONNECTION_TABLE block). Important note: It is very important to realize that the Hamiltonian depends on the a parameters used. Thus, when comparing relative energies for example, one has to take care that the a’s corresponding to the same bonds are identical.
Restarts¶
For the most part, restarts with ADF QM/MM are the same as in a standard ADF run. In other words to specify a restart, one needs to use the RESTART keyword. This signals the QM/MM extension to read the data from the QM/MM restart file as opposed to the input. It is important to emphasize that the QM/MM extension has its own restart file. Thus, the user will have to keep track to two restart files, the standard ADF restart file (TAPE21) and the QM/MM restart file. By default the QM/MM restart file is ‘mm.restart’, but you can change this by using the RESTART_FILE keyword within the QMMM key block. A truncated example of an ADF QM/MM restart input is given below:.
RESTART ADF_restart.file & *{same as in a standard ADF run}*
NOHESS
END
QMMM
RESTART_FILE mm_restart.file *{optional, with default filename of 'mm.restart'}*
FORCE_FIELD_FILE sybyl.ff
OUTPUT_LEVEL=1
WARNING_LEVEL=1
END
At the moment, when the RESTART keyword is used, the QM/MM extension will always look for the QM/MM restart file. There is no way to bypass this. However, the QM/MM restart file is simply a text file whose contents resemble those of the QMMM key block in the ADF QM/MM input and you can easily modify it.
It is also important to note that the RESTART_FILE both specifies the file to be read at the beginning of a restarted run, and the file that will be written to. In an ADF QM/MM run that was restarted, the initial QM/MM restart file is overwritten.
Geometry Constraints and Fixing Coordinates of MM atoms¶
Constraints can be applied to coordinates in the same way they are done with a standard ADF run (i.e. most commonly through the GEOVAR key block). There are no limitations to applying geometry constraints to QM or LI atoms, however, there exist some limitations to applying geometry constraints to MM atoms, particularly when using a Z-matrix.
When a geometry optimization is performed in Cartesian coordinates there is almost no limitation to applying constraints to MM atoms. One important exception is that linear transit constraints can not involve MM atoms. At the moment constraints cannot be applied to the coordinates that define the position of the MM atoms when internal (ZMAT) coordinates are used. Note that constraints still can be applied to the coordinates of the QM atoms, but just not to the MM atoms.
Symmetry constraints can not be applied with a QM/MM run, unless all of the MM atoms are frozen. Furthermore, the program will not check that the atoms of the MM region satisfy the imposed symmetry.
Miscellaneous Notes¶
Using ADF Dummy Atoms
It is often necessary to use dummy atoms when defining a Z-matrix for a geometry optimization. (Here we are not referring to the capping dummy atoms) Dummy atoms are allowed in a QM/MM simulation and they can be used just as they would in a normal ADF run. There are a few things to remember when using dummy atoms in a QM/MM run.
If a dummy atom is to be used to define the coordinates of QM atoms in a Z-matrix for geometry optimization, the dummy atom must precede any MM atoms in the atom list. In this case one should consider the dummy atom as part of the model QM system.
Atom should be given the Xx force field atom type in the MM_CONNECTION_TABLE key block. (In this way the atom is excluded from the non-bonded pair list)
Do not make any bonds to the dummy atom in the connection table; otherwise the program may require the user to define molecular mechanics potentials involving the dummy atom.
Dummy atoms are allowed in the MM region, however, they will only be used to define the initial coordinates. This is because the MM subsystem is always optimized in Cartesian space where dummy atoms are not necessary.
One can consider dummy atoms as part of the model QM system. For example, the following is not allowed because the dummy atom, which is assigned to the QM region, comes after atoms 5 and 6, which are MM, atoms.
...
MM_CONNECTION_TABLE
1 C_2 QM 2 3 4
2 O_2 QM 1
3 C_3 LI 1 8 9 10
4 C_3 QM 1 5 6 12
5 C_3 MM 4
6 C_3 MM 4
7 Xx QM
8 H MM 3
SUBEND
The example below is almost identical to the above example except that the dummy atom is assigned as a MM atom. Although this is allowed, this dummy atom will be optimized as a MM atom.
...
MM_CONNECTION_TABLE
1 C_2 QM 2 3 4
2 O_2 QM 1
3 C_3 LI 1 8 9 10
4 C_3 QM 1 5 6 12
5 C_3 MM 4
6 C_3 MM 4
7 Xx MM
8 H MM 3
SUBEND
The final example below, is probably what is wanted. Here the dummy atom is a QM atom and comes before any MM atoms. In this way, the dummy atom can be used to define the Z-matrix with the QM model system.
...
MM_CONNECTION_TABLE
1 C_2 QM 2 3 4
2 O_2 QM 1
3 C_3 LI 1 8 9 10
4 C_3 QM 1 5 6 12
5 Xx QM
6 C_3 MM 4
7 C_3 MM 4
8 H MM 3
SUBEND
Linear Transit Runs.
The QM/MM option can be used with the LINEAR TRANSIT option in ADF. However, the linear transit parameter or coordinate cannot involve any MM atoms. In other words, the linear transit parameter can only be defined in terms of QM atoms only.
Frequency Runs with QM/MM
At the moment, a FREQUENCY run can not be performed when using a QM/MM potential in ADF. This option has been implemented but has been disabled in this release for further testing.