2.6. ReaxFF: Adsorption on ZnS(110)¶
This tutorial illustrates ReaxFF parametrization for ZnS and H₂S on ZnS(110).
The tutorial teaches how to
- Combine several different force fields from the AMS ReaxFF library into a good initial guess
- Select training jobs to enter the training set
Note
The input files have already been prepared. If you simply want to run the
parametrization without preparing the input, copy the contents of
$AMSHOME/scripting/scm/params/examples/ZnS_ReaxFF to a new empty
directory.
To open the example in the graphical user interface (GUI): SCM → ParAMS,
File → Open the job_collection.yaml file in the new directory.
You can then run the parametrization.
2.6.1. Mix force fields from the AMS ReaxFF library¶
The ReaxFF force fields included with AMS cover a large part of the periodic table, however, far from all interactions are included.
In this tutorial, the goal is to model H₂S adsorption on a ZnS surface. No ZnS ReaxFF force field is included with AMS, but the following force fields might help:
| Force field | Chemistry | Reference |
| LiS.ff | Sulfur | Islam et al. |
| Mue2016.ff | Organic S-containing compounds | Müller and Hartke |
| ZnOH.ff | Zn and ZnO surfaces | Raymand et al. |
A ReaxFF force field contains parameters in the following categories: general (GEN), atomic (ATM), bond (BND), angle (ANG), off-diagonal (OFD), torsional (TOR), hydrogen bond (HBD).
Here, we’ll use the following initial parameters from the above force fields:
- LiS.ff: ATM:S, BND:S.S, ANG:S.S.S
- Mue2016.ff: ATM:H, BND:S.H, ANG:H.S.H
- ZnOH.ff: GEN, ATM:Zn, BND:H.H, BND:Zn.H, BND:Zn.Zn, BND:Zn.O*, ANG:Zn.Zn.O*, ANG:Zn.O*.Zn, ANG:Zn.O*.O*, ANG:O*.Zn.O*, HBD:O*.H.O*
For ZnOH.ff, the asterisks in e.g. BND:Zn.O* indicate that we will instead copy those parameters to initialize the BND:Zn.S block. That is, the Zn-O interaction in ZnOH.ff will be used as first guess for the Zn-S interaction in the new force field.
Here, we will demonstrate how to construct an initial ReaxFF force field from scratch. Open ParAMS in ReaxFF mode:
- SCM → ParAMSFile → NewParameters → ReaxFF
In the Parameters tab, you will see the ReaxFF parameters in the GEN block. Those parameters always exist for any ReaxFF force field.

We want to copy the GEN parameters from ZnOH.ff:
- Select one of the GEN parameters in the bottom table
- Parameters → Initialize ReaxFF block

- GEN: from
GEN:click OK.Note: this searches the ReaxFF force field library in AMS and can take a whileIn the resulting list, selectZnOH.ffand click OKThis updates the parameter values in the table

Now, add the ATM:S, BND:S.S, and ANG:S.S.S parameters from LiS.ff:
- Parameters → Add ReaxFF blockType
BND:S.S ANG:S.S.Sand click OK. This adds theBND:S.SandANG:S.S.Sblocks to the table, and also theATM:Sblock because it didn’t exist.The new parameters are automatically selected. Do not deselect them
- Parameters → Initialize ReaxFF blockANG:S.S.S from
ANG:S.S.S, ATM:S fromATM:S, BND:S.S fromBND:S.S. Click OKNote: this searches many ReaxFF force fields and can take a whileIn the list, select LiS.ff and click OKThis updates the parameters values in the table

Regularly save your work:
- File → SaveSave with the name
reaxff-zns.params
Next, add the ATM:H, BND:S.H and ANG:H.S.H from Mue2016.ff:
- Parameters → Add ReaxFF blockType
BND:S.H ANG:H.S.Hand click OK. This adds theBND:H.SandANG:H.S.Hblocks to the table, and also theATM:Hblock because it didn’t exist. The bond block is calledBND:H.S(sorted alphabetically).The new parameters are automatically selected. Do not deselect them

- Parameters → Initialize ReaxFF blockANG:H.S.H from
ANG:H.S.H, ATM:H fromATM:H, BND:H.S fromBND:H.S. Click OKNote: this searches many ReaxFF force fields and can take a whileIn the list, select Mue2016.ff and click OKFile → Save
Note
For the BND block the order of atoms do not matter (BND:H.S is the same as BND:S.H). For the ANG block the order does matter - the central atom is given in the middle, but for example ANG:H.S.Zn is equivalent to ANG:Zn.S.H
Now add the remaining blocks ATM:Zn, BND:H.H, BND:Zn.H, BND:Zn.Zn, BND:Zn.S, ANG:Zn.Zn.S, ANG:Zn.S.Zn, ANG:Zn.S.S, ANG:S.Zn.S
- Parameters → Add ReaxFF blockType
BND:H.H BND:Zn.H BND:Zn.Zn BND:Zn.S ANG:Zn.Zn.S ANG:Zn.S.Zn ANG:Zn.S.S ANG:S.Zn.S HBD:S.H.Sand click OK.The new parameters are automatically selected. Do not deselect them

- Parameters → Initialize ReaxFF blockReplace all
SwithOin the “from” fields. Click OK.

- Select ZnOH.ff in the list. Click OK.File → Save
This copies the values from the BND:Zn.O block in ZnOH.ff to the BND:Zn.S block in your new force field, copies the values from the ANG:Zn.Zn.O block in ZnOH.ff to the ANG:Zn.Zn.S block in your new force field, etc.
This is accomplished in the get_initial_parameters() method of generate_input_files.py:
def get_initial_parameters(bounds_scale=1.3):
LiS = ReaxFFParameters('LiS.ff', bounds_scale=bounds_scale) # http://dx.doi.org/10.1039/C4CP04532G
Mue2016 = ReaxFFParameters('Mue2016.ff', bounds_scale=bounds_scale) # http://dx.doi.org/10.1021/acs.jctc.6b00461
ZnOH = ReaxFFParameters('ZnOH.ff', bounds_scale=bounds_scale) # http://dx.doi.org/10.1016/j.susc.2009.12.012
# interf is the new parameter interface
interf = ReaxFFParameters(None)
# copy S parameters from LiS
interf.copy_block(LiS, 'ATM:S')
interf.copy_block(LiS, 'BND:S.S')
interf.copy_block(LiS, 'ANG:S.S.S')
# copy H, S.H, and H.S.H parameters from Mue2016
interf.copy_block(Mue2016, 'ATM:H')
interf.copy_block(Mue2016, 'BND:S.H')
interf.copy_block(Mue2016, 'ANG:H.S.H')
# copy the GEN block and many other blocks from ZnOH, replacing O with S
interf.copy_block(ZnOH, 'GEN')
interf.copy_block(ZnOH, 'ATM:Zn')
interf.copy_block(ZnOH, 'BND:H.H', 'BND:H.H')
interf.copy_block(ZnOH, 'BND:Zn.H', 'BND:Zn.H')
interf.copy_block(ZnOH, 'BND:Zn.Zn', 'BND:Zn.Zn')
interf.copy_block(ZnOH, 'BND:Zn.O', 'BND:Zn.S')
interf.copy_block(ZnOH, 'ANG:Zn.Zn.O', 'ANG:Zn.Zn.S')
interf.copy_block(ZnOH, 'ANG:Zn.O.Zn', 'ANG:Zn.S.Zn')
interf.copy_block(ZnOH, 'ANG:Zn.O.O', 'ANG:Zn.S.S')
interf.copy_block(ZnOH, 'ANG:O.Zn.O', 'ANG:S.Zn.S')
interf.copy_block(ZnOH, 'HBD:O.H.O', 'HBD:S.H.S')
# give a title
interf.header['head'] = 'ZnS parametrization'
return interf
2.6.2. Choose the parameters to optimize¶
We will not optimize (deactivate)
- Any GEN (general) parameters
- Any ATM (atomic) parameters
- Any parameters with category
DoNotOptimize(these should never be optimized) - Any BND (bond) parameters for H-Zn or H-H (there is no training data for those interactions)
- Any BND (bond) parameters for π-bonds (there is no training data)
- Any ANG (angle) parameters for S-S-S (there is no training data)
- Any parameters with value 0 (usually means “off”)
We will optimize (activate):
| Block | Atoms | Parameters |
| BND | S-S, H-S, Zn-Zn, S-Zn | D_e^sigma p_be1 p_ovun1 p_be2 p_bo1 p_bo2 |
| ANG | H-S-H, S-Zn-Zn, Zn-S-Zn, S-S-Zn, S-Zn-S | Theta_0,0 p_val1 p_val2 p_val7 p_val4 |
| HBD | S-H-S | r_hb^0 p_hb1 -p_hb2 -p_hb3 |
There are various ways to select the above list of parameters for optimization.
For example, the BND parameters can be selected as follows:
- Switch to the Parameters tabIn the search box at the bottom, type
S.S:In the Block dropdown, select BNDIn the Category dropdown, select StandardTick the Active checkbox forD_e^sigmap_be1p_ovun1p_be2p_bo1p_bo2
- Repeat the above when searching for
H.S:,Zn.Zn:, andS.Zn:Save your work before continuing
The ANG parameters can be selected as follows:
- In the Block dropdown, select ANGIn the Category dropdown, select AllIn the search box at the bottom, type
thetaTick the Active checkbox for H.S.H, S.S.Zn, S.Zn.S, S.Zn.Zn, Zn.S.Zn (but not for S.S.S)

- Repeat the above steps searching for
p_val1,p_val2,``p_val7``,p_val4Save your work before continuing
The HBD parameters can be selected as follows:
- In the Block dropdown, select HBDIn the Category dropdown, select AllTick the Active checkbox for all S.H.S HBD parameters.Save your work before continuing

Verify that you’ve activated the desired parameters:
- In the Block dropdown, select AllIn the Category dropdown, select AllIn the Active dropdown, unselect Show Non-Active

You should now see only the active parameters.
By default, the Min and the Max values are set to ±20% of the value. Let’s increase the range for all the now active parameters by another 8.3%:
- Select the first active parameter (
S.S:D_e^sigma)Scroll down to the bottomHold SHIFT and click on the last active parameter (S.H.S:-p_hb3)

- In the Parameters menu, select Adjust Min and Max
- In the left drop-down, select RangeIn the middle drop-down, select ScaleSet the value to 1.083Click OK

This increases the range (decreases Min and increases Max) for all selected parameters by 8.3%. In the Adjust Min and Max dialog, you can Scale/Shift/Set the Range/Min/Max, for many selected parameters at the same time.
Save your work before continuing:
- File → Save
The function activate_parameters() in generate_input_files.py
applies some logic to decide whether to activate a parameter during the
parametrization:
def activate_parameters(interf: ReaxFFParameters):
# only optimize bond and angle parameters
# do not optimize H-Zn and H-H bond parameters
# do not optimize pi-bonding parameters
# parameters with a value of 0 are usually best kept at 0
for p in interf:
if p.metadata['category'] == 'DoNotOptimize':
p.is_active = False
elif p.metadata['block'] == 'BND':
p.is_active = 'n/a' not in p.name.lower() and \
not p.name.endswith('pi') and \
p.value != 0 and \
p.metadata['atoms'] != ['H', 'Zn'] and \
p.metadata['atoms'] != ['Zn', 'H'] and \
p.metadata['atoms'] != ['H', 'H'] and \
not 'pi bond' in p.metadata['description'].lower()
elif p.metadata['block'] == 'ANG':
p.is_active = 'n/a' not in p.name.lower() and \
p.value != 0 and \
p.metadata['atoms'] != ['S', 'S', 'S']
elif p.metadata['block'] == 'HBD':
p.is_active = True
else:
p.is_active = False
For the BND parameters, we choose not to optimize any parameters that should always be constant(with category DoNotOptimize), the π-bond parameters, any parameters with value 0, or the Zn-H and H-H parameters.
Similar conditions are placed for the ANG parameters.
2.6.3. ZnS training and validation set¶
The training data contains
Polymorphs
- Energy-volume scans of the zinc blende and rock salt polymorphs of ZnS (wurtzite energy-volume scan in the validation set). See the GFN1-xTB tutorial for how to set up an energy-volume scan reference calculation in AMS and import it into ParAMS.
- Enthalpy of formation of the zinc blende, wurtzite, and rocksalt polymorphs of ZnS
Gaseous H₂S
- Optimized S-H bond length in H₂S, and H-S-H angle. See the GFN1-xTB tutorial or Import training data (GUI) tutorial for how to import the bond length from a geometry optimization job.
- Bond scan for the S-H bond and angle scan for the H-S-H angle in gaseous H₂S. See the ReaxFF: Gaseous H₂O tutorial for how to set up a bond scan reference calculation in AMS.
Clean ZnS(110) surface
- Optimized bond lengths and angles at a clean ZnS(110) surface. See also: Building Crystals and Slabs and Crystals and Surfaces
- Formation energy (surface energy) of the ZnS(110) surface. See also: Surface energy
- Forces on distorted ZnS(110) surfaces (*)
H₂S adsorbed on ZnS(110)
- Optimized Zn-S and H…S bond lengths for H₂S/ZnS(110)
- Adsorption energy of H₂S/ZnS(110). See also: Adsorption
Other
- Atomic charges for a variety of systems
(*): The distorted ZnS(110) surfaces were obtained by geometry-optimizing the clean ZnS(110) surface using a previous iteration of the force field.
Note
This tutorial does not cover how to set up all the reference calculations. In the GUI, if you switch to the Jobs panel and double-click in the Detail column, you can use the button Edit in AMSinput to see how the reference calculation was set up (approximately, some details like the maximum number of iterations for the geometry optimization may differ).
If you have run a reference calculation, in the ParAMS GUI you can choose Jobs → Add Job, and browse to the ams.rkf file from the job. That will let you import many properties (energy, forces, stress tensor, charges, …).
The needed .yaml files can be found in
$AMSHOME/scripting/scm/params/examples/ZnS_ReaxFF, where
$AMSHOME is the AMS installation directory.
Note
Using File → Merge you can add jobs, training set entries, and engines into the current project. If you select File → Open, you will start a new project (thus losing the parameters that you set up in the previous section!).
- File → Merge → Add to Jobs. Select the
job_collection.yamlfile.File → Merge → Add to training set. Select thetraining_set.yamlfile.File → Merge → Add to training set. Select thevalidation_set.yamlfile.File → Merge → Add to Engines. Select thejob_collection_engines.yamlfile.

First download and unzip reference_calculation_results.zip.
It contains trimmed-down results files from BAND DFT calculations.
Then take a look at the import_results() function in generate_input_files.py.
It uses a Results Importer to import results from different types of DFT reference calculations, for example PES Scans and geometry optimizations.
def import_results():
ri = ResultsImporter(settings={'units': {'energy': 'eV', 'forces': 'eV/angstrom'}, 'remove_bonds': True})
# energy-volume scans
ri.add_singlejob('dft/ZnS_pesscans/zincblende/ams.rkf', task='PESScan', properties={'pes': {'weight': 5.0}})
ri.add_singlejob('dft/ZnS_pesscans/rocksalt/ams.rkf', task='PESScan', properties={'pes': {'weight': 4.0}})
ri.add_singlejob('dft/ZnS_pesscans/wurtzite/ams.rkf', task='PESScan', properties='pes', data_set='validation_set')
# single-points
ri.add_singlejob('dft/ZnS_pesscans/wurtzite_sp/ams.rkf', properties='charges')
ri.add_singlejob('dft/ZnS_pesscans/rocksalt_sp/ams.rkf', properties='charges')
ri.add_singlejob('dft/ZnS_pesscans/zincblende_sp/ams.rkf', properties='charges')
# optimized bond lengths, charges, gasphase H2S
ri.add_singlejob('dft/band_h2s.results/ams.rkf', task='GeometryOptimization', properties=['distance(0,1)', 'angle(1,0,2)', 'charges'])
# bond and angle scans, gasphase H2S
ri.add_singlejob('dft/bondscan_h2s_pbesol.results/ams.rkf', task='PESScan', properties={'pes': {'weight': 3.0}})
ri.add_singlejob('dft/anglescan_h2s_pbesol.results/ams.rkf', task='PESScan', properties='pes')
# H2S adsorbed on ZnS(110), bond lengths and charges
ri.add_singlejob('dft/band_h2s_110.results/ams.rkf', task='GeometryOptimization', properties=['distance(34,19)', 'distance(14, 37)', 'charges'])
# enthalpy of formations
ri.add_reaction_energy(reactants=['dft/sulfur.results', 'dft/Zn.results'], products=['dft/ZnS_pesscans/wurtzite_sp'], normalization='p0', normalization_value=0.5)
ri.add_reaction_energy(reactants=['dft/sulfur.results', 'dft/Zn.results'], products=['dft/ZnS_pesscans/rocksalt_sp'], normalization='p0', normalization_value=1.0)
ri.add_reaction_energy(reactants=['dft/sulfur.results', 'dft/Zn.results'], products=['dft/ZnS_pesscans/zincblende_sp'], normalization='p0', normalization_value=1.0)
# relative polymorph energies
ri.add_reaction_energy(reactants=['dft/ZnS_pesscans/wurtzite_sp'], products=['dft/ZnS_pesscans/zincblende_sp'], normalization='p0', normalization_value=1.0)
ri.add_reaction_energy(reactants=['dft/ZnS_pesscans/rocksalt_sp'], products=['dft/ZnS_pesscans/zincblende_sp'], normalization='p0', normalization_value=1.0)
# adsorption energy
ri.add_reaction_energy(reactants=['dft/band_110.results', 'dft/band_h2s.results'], products=['dft/band_h2s_110.results/ams.rkf'], normalization='p0', normalization_value=1.0, task='GeometryOptimization')
# "surface energy", in a way
ri.add_reaction_energy(reactants=['wurtzite_sp'], products=['dft/band_110_noconstraints.results'], normalization='r0', normalization_value=0.5, task='GeometryOptimization')
ri.add_singlejob('dft/band_110_noconstraints.results', task='GeometryOptimization', properties=['distance(39,42)', 'distance(19, 42)', 'distance(42,47)', 'distance(27,47)', 'distance(4,47)', 'distance(4,12)', 'distance(3,12)', 'distance(12,15)', 'distance(5,28)', 'angle(42,39,19)', 'angle(39,19,31)', 'angle(39,19,24)', 'angle(39,42,47)', 'angle(23,27,47)', 'angle(27,47,4)', 'dihedral(9,28,27,23)', 'charges'])
# some singlepoints
ri.add_singlejob('dft/band_distorted_ads_110.results/', properties='forces')
ri.add_singlejob('dft/band_distorted_clean_110.results', properties='forces')
ri.add_reaction_energy(reactants=['dft/band_distorted_ads_110.results'], products=['band_h2s_110'], sigma=2.0)
ri.add_reaction_energy(reactants=['dft/band_distorted_clean_110.results'], products=['band_110'], sigma=2.0)
# save training_set.yaml, validation_set.yaml, job_collection.yaml
ri.store(backup=False, binary=False)
2.6.4. Run the ZnS ReaxFF parametrization¶
- File → Save As with a new nameFile → Run (or, select the job in AMSjobs, set a remote queue, and submit to a compute cluster)
Run
"$AMSBIN/params" optimize
2.6.5. ZnS parametrization results¶
Note
These results only give an example of a possible result. If you rerun the parametrization, your results will likely differ and could either be better or worse.

2.6.5.1. ZnS energies¶
| Reference (eV) | Prediction (eV) | |
| ZnS enthalpy of formation (zincblende) | -2.737 | -2.737 |
| ZnS enthalpy of formation (rock salt) | -2.329 | -2.360 |
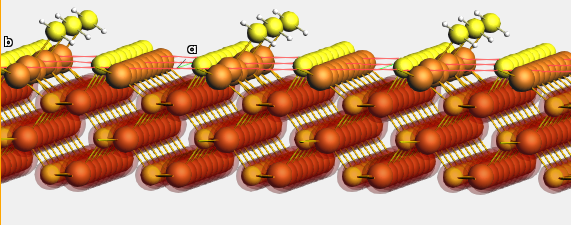
| H₂S adsorption energy on ZnS(110) (a) | -0.742 | -0.559 |
| ZnS(110) slab energy relative to bulk (b) | +0.124 | +0.257 |
(a): This adsorption energy was not calculated using the in-cell approach
(b): This energy can be converted into a surface energy


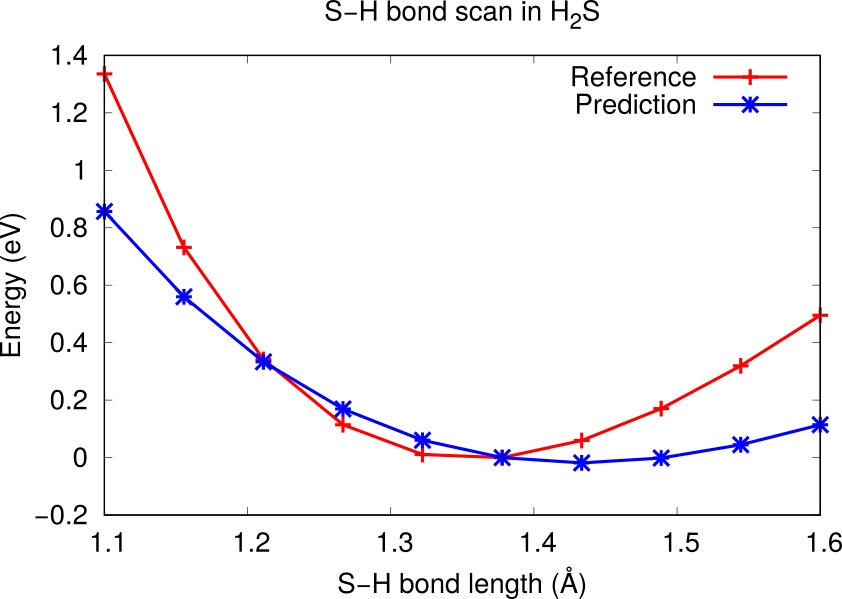

2.6.5.4. ZnS optimized distances¶

2.6.5.5. ZnS forces on distorted structures¶

Fig. 2.4 Reference and predicted forces on the two depicted structures. The structures were obtained by geometry-optimizing a slab using a previous iteration of the force field.