Transition state search for polymerization: Ziegler Natta catalyst¶
See the corresponding AMS GUI tutorial for background information and how to perform these steps with the graphical user interface.
To follow along, either
Download
ZieglerNattaCatalyst.py(run as$AMSBIN/amspython ZieglerNattaCatalyst.py).Download
ZieglerNattaCatalyst.ipynb(see also: how to install Jupyterlab in AMS)
Worked Example¶
Plan¶
This tutorial shows how to
optimize the reactant state,
perform a PES Scan to get a good starting structure for transition state search,
perform a transition state search,
extract the barrier
Note that this tutorial only shows one half of a reaction.
Initial imports¶
from scm.base import ChemicalSystem
from scm.plams import view, Settings, AMSJob, Units
Initial system¶
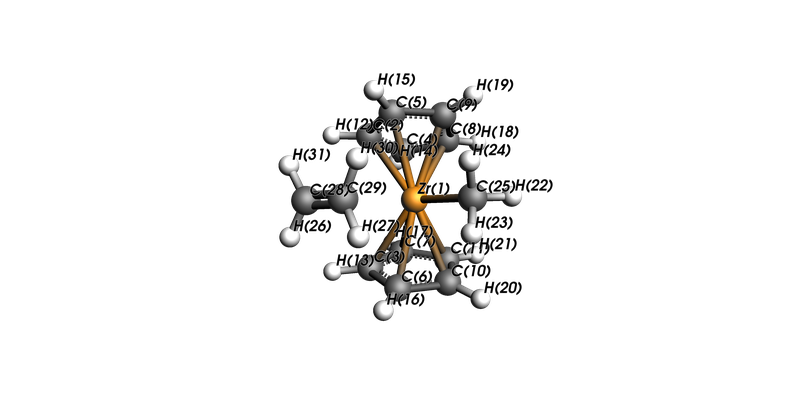
mol = ChemicalSystem(
"""
System
Charge 1.0
Atoms
Zr 1.60682759 -9.09697636 1.29667400
C 2.89496678 -10.06750666 3.11673910
C 2.84805610 -9.90469975 -0.63831357
C 1.91952398 -11.03393562 2.76898899
C 2.21720521 -8.93716871 3.64709384
C 2.05919385 -8.80831375 -1.07706558
C 1.96898612 -10.95146163 -0.26897070
C 0.63990901 -10.49043590 3.04466191
C 0.82713373 -9.20286661 3.61113029
C 0.69607878 -9.17611676 -0.96529901
C 0.63812917 -10.49403820 -0.44270184
H 3.96827494 -10.23973626 3.15013890
H 3.92582990 -9.99794535 -0.74784951
H 2.12190386 -12.06174102 2.48123664
H 2.68569500 -8.09238646 4.14547290
H 2.43329579 -7.91168000 -1.56456659
H 2.26368032 -11.97103749 -0.03692581
H -0.30289388 -11.03081473 2.99676260
H 0.05404650 -8.59747583 4.07520644
H -0.14620660 -8.61151792 -1.35464113
H -0.25838864 -11.10457655 -0.36003217
H -0.85662057 -7.76723654 1.38227139
H 0.26321351 -6.74399730 0.47215750
H 0.31659006 -6.75672110 2.23713123
C 0.17801499 -7.39347280 1.35339060
H 4.83158681 -7.79302616 0.33628574
H 2.98632816 -6.14645123 0.41958259
C 4.37932750 -7.48955975 1.27484960
C 3.37901781 -6.61143075 1.32052856
H 3.01865674 -6.20459134 2.26225286
H 4.86414560 -7.85327440 2.17514370
End
End"""
)
mol.guess_bonds()
print(f"{mol.charge=}")
view(mol, direction="along_y", show_atom_labels=True, atom_label_type="Name")
mol.charge=1.0
DFTB Settings¶
from scm.plams import Settings, AMSJob
def get_dftb_settings():
# Engine settings: GFN-1xTB with implicit Tolene solvation
settings = Settings()
settings.input.DFTB.Model = "GFN1-xTB"
settings.input.DFTB.Solvation.Solvent = "Toluene"
return settings
print(AMSJob(settings=get_dftb_settings()).get_input())
Engine DFTB
Model GFN1-xTB
Solvation
Solvent Toluene
End
EndEngine
Geometry optimization of reactant state¶
def get_reactants_job(mol):
settings = get_dftb_settings()
settings.input.ams.Task = "SinglePoint"
settings.input.ams.Properties.NormalModes = "Yes"
job = AMSJob(molecule=mol, settings=settings, name="reactants_frequencies")
return job
reactants_job = get_reactants_job(mol)
reactants_job.run();
[16.12|15:06:37] JOB reactants_frequencies STARTED
[16.12|15:06:37] JOB reactants_frequencies RUNNING
[16.12|15:06:40] JOB reactants_frequencies FINISHED
[16.12|15:06:40] JOB reactants_frequencies SUCCESSFUL
Set up PES Scan to find approximate transition state¶
def get_pesscan_job(mol):
# Engine settings: GFN-1xTB with implicit Tolene solvation
settings = get_dftb_settings()
# AMS settings for PES scan (see online tutorial for choice of scan coordinates)
settings.input.ams.Task = "PESScan"
settings.input.ams.PESScan.ScanCoordinate.nPoints = "20"
settings.input.ams.PESScan.ScanCoordinate.Distance = ["1 28 3.205 2.3", "29 25 3.295 1.5"]
# Loosened convergence criteria
settings.input.ams.GeometryOptimization.Convergence.Energy = "5.0e-5 [Hartree]"
settings.input.ams.GeometryOptimization.Convergence.Gradients = "5.0e-3"
settings.input.ams.GeometryOptimization.Convergence.Step = "5.0e-3 [Angstrom]"
# setting up the AMS job with the molecule and settings object
job = AMSJob(molecule=mol, settings=settings, name="pes_scan")
return job
# run the calulation
pesscan_job = get_pesscan_job(reactants_job.results.get_main_system())
pesscan_job.run();
[16.12|15:06:40] JOB pes_scan STARTED
[16.12|15:06:40] JOB pes_scan RUNNING
[16.12|15:07:13] JOB pes_scan FINISHED
[16.12|15:07:13] JOB pes_scan SUCCESSFUL
PESScan results¶
import matplotlib.pyplot as plt
from pprint import pprint
pesscan_results = pesscan_job.results.get_pesscan_results()
pprint(pesscan_results, depth=1)
{'ConstrainedAtoms': {1, 28, 29, 25},
'Converged': [...],
'HistoryIndices': [...],
'Molecules': [...],
'OrigScanCoords': [...],
'PES': [...],
'Properties': [],
'RaveledPESCoords': [...],
'RaveledScanCoords': [...],
'RaveledUnits': [...],
'ScanCoords': [...],
'Units': [...],
'nPESPoints': 20,
'nRaveledScanCoords': 2,
'nScanCoords': 1}
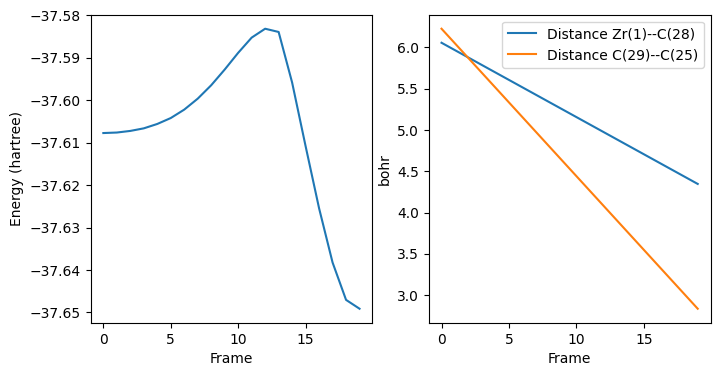
fix, ax = plt.subplots(1, 2, figsize=(8, 4))
ax[0].plot(list(range(pesscan_results["nPESPoints"])), pesscan_results["PES"])
ax[0].set_ylabel("Energy (hartree)")
ax[0].set_xlabel("Frame")
for i in range(pesscan_results["nRaveledScanCoords"]):
ax[1].plot(
list(range(pesscan_results["nPESPoints"])),
pesscan_results["RaveledPESCoords"][i],
label=pesscan_results["RaveledScanCoords"][i],
)
ax[1].set_ylabel("bohr")
ax[1].set_xlabel("Frame")
ax[1].legend();
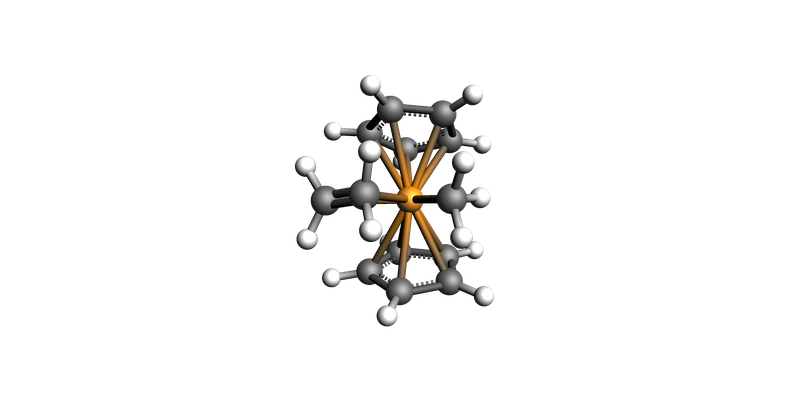
import numpy as np
highest_energy = np.max(pesscan_results["PES"])
highest_energy_index = np.argmax(pesscan_results["PES"])
# res['Molecules'] contains all the converged geometries (as PLAMS Molecules)
highest_energy_geo = pesscan_results["Molecules"][highest_energy_index]
print(f"Highest energy: {highest_energy:.6f} hartree, for geometry #{highest_energy_index+1}")
print("We will use that structure as starting point for TS search")
highest_energy_geo.guess_bonds()
view(highest_energy_geo, direction="along_y")
Highest energy: -37.583147 hartree, for geometry #13
We will use that structure as starting point for TS search
Transition state search¶
def get_ts_job(mol):
settings = get_dftb_settings()
# AMS settings for TS search
settings.input.ams.Task = "TransitionStateSearch"
settings.input.ams.Properties.NormalModes = "Yes"
settings.input.ams.GeometryOptimization.InitialHessian.Type = "Calculate"
# setting up the AMS job with the molecule and settings object
job = AMSJob(molecule=mol, settings=settings, name="ts_search")
return job
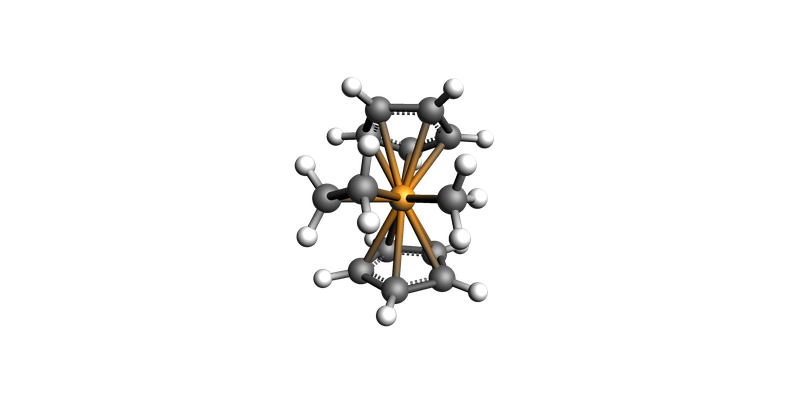
ts_job = get_ts_job(highest_energy_geo)
ts_job.run()
ts_mol = ts_job.results.get_main_system()
ts_mol.guess_bonds()
view(ts_mol, direction="along_y")
[16.12|15:07:15] JOB ts_search STARTED
[16.12|15:07:15] JOB ts_search RUNNING
[16.12|15:07:26] JOB ts_search FINISHED
[16.12|15:07:26] JOB ts_search SUCCESSFUL
def get_gibbs_energy(job: AMSJob, unit="hartree"):
gibbs_energy = job.results.readrkf("Thermodynamics", "Gibbs free Energy", file="engine")
gibbs_energy *= Units.convert(1.0, "hartree", unit)
return gibbs_energy
def print_ts_results(job):
energy = job.results.get_energy(unit="kcal/mol")
gibbs_energy = get_gibbs_energy(job, unit="kcal/mol")
frequencies = job.results.get_frequencies(unit="cm^-1")
character = job.results.readrkf("AMSResults", "PESPointCharacter", file="engine")
# print results
print("== Results ==")
print("Character : {}".format(character))
print("Energy : {:.3f} kcal/mol".format(energy))
print("Gibbs Energy: {:.3f} kcal/mol".format(gibbs_energy))
for freq in frequencies:
if freq < 0:
print("Imaginary frequency : {:.3f} cm^-1".format(freq))
else:
break
print_ts_results(ts_job)
== Results ==
Character : transition state
Energy : -23587.067 kcal/mol
Gibbs Energy: -23460.913 kcal/mol
Imaginary frequency : -397.387 cm^-1
Free energy barrier¶
def print_final_results(gibbs_energy_ts: float, gibbs_energy_reactants: float, T=298.15):
barrier = gibbs_energy_ts - gibbs_energy_reactants # kJ/mol
NA = 6.022e23 # Avogadro's constant
k_B = 1.380649e-23 # Boltzmann constant, J / (mol*K)
h = 6.626e-34 # Planck's constant, J * s
rate_constant = (k_B * T / h) * np.exp(-barrier * 1000 / (NA * k_B * T))
print("Results summary")
print("Reactants Gibbs Energy: {:.3f} kJ/mol".format(gibbs_energy_reactants))
print("TS Gibbs Energy: {:.3f} kJ/mol".format(gibbs_energy_ts))
print("Free energy barrier: {:.3f} kJ/mol".format(barrier))
print("Rate constant: {:.3f} s⁻¹ at {} K".format(rate_constant, T))
gibbs_energy_ts = get_gibbs_energy(ts_job, "kJ/mol")
gibbs_energy_reactants = get_gibbs_energy(reactants_job, "kJ/mol")
print_final_results(gibbs_energy_ts, gibbs_energy_reactants, T=298.15)
Results summary
Reactants Gibbs Energy: -98216.185 kJ/mol
TS Gibbs Energy: -98160.461 kJ/mol
Free energy barrier: 55.724 kJ/mol
Rate constant: 1073.251 s⁻¹ at 298.15 K
Complete Python code¶
#!/usr/bin/env amspython
# coding: utf-8
# ## Plan
#
# This tutorial shows how to
#
# * optimize the reactant state,
# * perform a PES Scan to get a good starting structure for transition state search,
# * perform a transition state search,
# * extract the barrier
#
# Note that this tutorial only shows one half of a reaction.
# ## Initial imports
from scm.base import ChemicalSystem
from scm.plams import view, Settings, AMSJob, Units
# ## Initial system
mol = ChemicalSystem(
"""
System
Charge 1.0
Atoms
Zr 1.60682759 -9.09697636 1.29667400
C 2.89496678 -10.06750666 3.11673910
C 2.84805610 -9.90469975 -0.63831357
C 1.91952398 -11.03393562 2.76898899
C 2.21720521 -8.93716871 3.64709384
C 2.05919385 -8.80831375 -1.07706558
C 1.96898612 -10.95146163 -0.26897070
C 0.63990901 -10.49043590 3.04466191
C 0.82713373 -9.20286661 3.61113029
C 0.69607878 -9.17611676 -0.96529901
C 0.63812917 -10.49403820 -0.44270184
H 3.96827494 -10.23973626 3.15013890
H 3.92582990 -9.99794535 -0.74784951
H 2.12190386 -12.06174102 2.48123664
H 2.68569500 -8.09238646 4.14547290
H 2.43329579 -7.91168000 -1.56456659
H 2.26368032 -11.97103749 -0.03692581
H -0.30289388 -11.03081473 2.99676260
H 0.05404650 -8.59747583 4.07520644
H -0.14620660 -8.61151792 -1.35464113
H -0.25838864 -11.10457655 -0.36003217
H -0.85662057 -7.76723654 1.38227139
H 0.26321351 -6.74399730 0.47215750
H 0.31659006 -6.75672110 2.23713123
C 0.17801499 -7.39347280 1.35339060
H 4.83158681 -7.79302616 0.33628574
H 2.98632816 -6.14645123 0.41958259
C 4.37932750 -7.48955975 1.27484960
C 3.37901781 -6.61143075 1.32052856
H 3.01865674 -6.20459134 2.26225286
H 4.86414560 -7.85327440 2.17514370
End
End"""
)
mol.guess_bonds()
print(f"{mol.charge=}")
view(mol, direction="along_y", show_atom_labels=True, atom_label_type="Name")
# ## DFTB Settings
from scm.plams import Settings, AMSJob
def get_dftb_settings():
# Engine settings: GFN-1xTB with implicit Tolene solvation
settings = Settings()
settings.input.DFTB.Model = "GFN1-xTB"
settings.input.DFTB.Solvation.Solvent = "Toluene"
return settings
print(AMSJob(settings=get_dftb_settings()).get_input())
# ## Geometry optimization of reactant state
def get_reactants_job(mol):
settings = get_dftb_settings()
settings.input.ams.Task = "SinglePoint"
settings.input.ams.Properties.NormalModes = "Yes"
job = AMSJob(molecule=mol, settings=settings, name="reactants_frequencies")
return job
reactants_job = get_reactants_job(mol)
reactants_job.run()
# ## Set up PES Scan to find approximate transition state
def get_pesscan_job(mol):
# Engine settings: GFN-1xTB with implicit Tolene solvation
settings = get_dftb_settings()
# AMS settings for PES scan (see online tutorial for choice of scan coordinates)
settings.input.ams.Task = "PESScan"
settings.input.ams.PESScan.ScanCoordinate.nPoints = "20"
settings.input.ams.PESScan.ScanCoordinate.Distance = ["1 28 3.205 2.3", "29 25 3.295 1.5"]
# Loosened convergence criteria
settings.input.ams.GeometryOptimization.Convergence.Energy = "5.0e-5 [Hartree]"
settings.input.ams.GeometryOptimization.Convergence.Gradients = "5.0e-3"
settings.input.ams.GeometryOptimization.Convergence.Step = "5.0e-3 [Angstrom]"
# setting up the AMS job with the molecule and settings object
job = AMSJob(molecule=mol, settings=settings, name="pes_scan")
return job
# run the calulation
pesscan_job = get_pesscan_job(reactants_job.results.get_main_system())
pesscan_job.run()
# ## PESScan results
import matplotlib.pyplot as plt
from pprint import pprint
pesscan_results = pesscan_job.results.get_pesscan_results()
pprint(pesscan_results, depth=1)
fix, ax = plt.subplots(1, 2, figsize=(8, 4))
ax[0].plot(list(range(pesscan_results["nPESPoints"])), pesscan_results["PES"])
ax[0].set_ylabel("Energy (hartree)")
ax[0].set_xlabel("Frame")
for i in range(pesscan_results["nRaveledScanCoords"]):
ax[1].plot(
list(range(pesscan_results["nPESPoints"])),
pesscan_results["RaveledPESCoords"][i],
label=pesscan_results["RaveledScanCoords"][i],
)
ax[1].set_ylabel("bohr")
ax[1].set_xlabel("Frame")
ax[1].legend()
import numpy as np
highest_energy = np.max(pesscan_results["PES"])
highest_energy_index = np.argmax(pesscan_results["PES"])
# res['Molecules'] contains all the converged geometries (as PLAMS Molecules)
highest_energy_geo = pesscan_results["Molecules"][highest_energy_index]
print(f"Highest energy: {highest_energy:.6f} hartree, for geometry #{highest_energy_index+1}")
print("We will use that structure as starting point for TS search")
highest_energy_geo.guess_bonds()
view(highest_energy_geo, direction="along_y")
# ## Transition state search
def get_ts_job(mol):
settings = get_dftb_settings()
# AMS settings for TS search
settings.input.ams.Task = "TransitionStateSearch"
settings.input.ams.Properties.NormalModes = "Yes"
settings.input.ams.GeometryOptimization.InitialHessian.Type = "Calculate"
# setting up the AMS job with the molecule and settings object
job = AMSJob(molecule=mol, settings=settings, name="ts_search")
return job
ts_job = get_ts_job(highest_energy_geo)
ts_job.run()
ts_mol = ts_job.results.get_main_system()
ts_mol.guess_bonds()
view(ts_mol, direction="along_y")
def get_gibbs_energy(job: AMSJob, unit="hartree"):
gibbs_energy = job.results.readrkf("Thermodynamics", "Gibbs free Energy", file="engine")
gibbs_energy *= Units.convert(1.0, "hartree", unit)
return gibbs_energy
def print_ts_results(job):
energy = job.results.get_energy(unit="kcal/mol")
gibbs_energy = get_gibbs_energy(job, unit="kcal/mol")
frequencies = job.results.get_frequencies(unit="cm^-1")
character = job.results.readrkf("AMSResults", "PESPointCharacter", file="engine")
# print results
print("== Results ==")
print("Character : {}".format(character))
print("Energy : {:.3f} kcal/mol".format(energy))
print("Gibbs Energy: {:.3f} kcal/mol".format(gibbs_energy))
for freq in frequencies:
if freq < 0:
print("Imaginary frequency : {:.3f} cm^-1".format(freq))
else:
break
print_ts_results(ts_job)
# ## Free energy barrier
def print_final_results(gibbs_energy_ts: float, gibbs_energy_reactants: float, T=298.15):
barrier = gibbs_energy_ts - gibbs_energy_reactants # kJ/mol
NA = 6.022e23 # Avogadro's constant
k_B = 1.380649e-23 # Boltzmann constant, J / (mol*K)
h = 6.626e-34 # Planck's constant, J * s
rate_constant = (k_B * T / h) * np.exp(-barrier * 1000 / (NA * k_B * T))
print("Results summary")
print("Reactants Gibbs Energy: {:.3f} kJ/mol".format(gibbs_energy_reactants))
print("TS Gibbs Energy: {:.3f} kJ/mol".format(gibbs_energy_ts))
print("Free energy barrier: {:.3f} kJ/mol".format(barrier))
print("Rate constant: {:.3f} s⁻¹ at {} K".format(rate_constant, T))
gibbs_energy_ts = get_gibbs_energy(ts_job, "kJ/mol")
gibbs_energy_reactants = get_gibbs_energy(reactants_job, "kJ/mol")
print_final_results(gibbs_energy_ts, gibbs_energy_reactants, T=298.15)