Conformers Generation with Multiple Molecules¶
See also
Conformers PLAMS interface
Conformers Generation PLAMS example
Example illustrating how to generate conformers with AMS.
This example is compatible with AMS2024+.
To follow along, either
Download
conformers.py(run as$AMSBIN/amspython conformers.py).Download
conformers.ipynb(see also: how to install Jupyterlab in AMS)
Worked Example¶
Lets see how two alanine molecules orient themselves using CREST conformer generation. To do this we will constrain the system in a spherical region using the SphericalWall constraint. We start by setting up a system of two alanine molecules in a relatively small space.
Initial imports¶
import scm.plams as plams
import sys
from scm.conformers import ConformersJob
import numpy as np
import matplotlib.pyplot as plt
import os
try:
from scm.plams import view # view molecule using AMSview in a Jupyter Notebook in AMS2026+
_has_view = True
except ImportError:
from scm.plams import plot_molecule # plot molecule in a Jupyter Notebook in AMS2023+
_has_view = False
def view(molecule, ax=None, **kwargs):
plot_molecule(molecule, ax=ax)
# this line is not required in AMS2025+
plams.init();
PLAMS working folder: /path/plams/examples/ConformersMultipleMolecules/plams_workdir
Single alanine molecule¶
smiles = "CC(N)C(=O)O"
alanine = plams.from_smiles(smiles)
view(alanine, height=300, width=300)
Initial system: alanine dimer¶
Pack two alanine molecules in a sphere with a density of 0.5 kg/L.
density = 0.5
mol = plams.packmol(alanine, n_molecules=2, density=density, sphere=True)
Translate the molecule to be centered around the origin (needed for SphericalWall later):
mol.translate(-np.array(mol.get_center_of_mass()))
view(mol, direction="along_pca3")

Calculation setup¶
To determine the radius of the SphericalWall we measure the size of the initial dimer.
dists = plams.distance_array(mol, mol)
max_dist = np.max(dists)
diameter = 1.33 * max_dist
radius = diameter / 2
print(f"Largest distance between atoms: {max_dist:.3f} ang.")
print(f"Radius: {radius:.3f} ang.")
Largest distance between atoms: 8.361 ang.
Radius: 5.560 ang.
Now we can set up the Crest conformer generation job, with the appropriate spherical wall constraining the molecules close together.
settings = plams.Settings()
settings.input.ams.EngineAddons.WallPotential.Enabled = "Yes"
settings.input.ams.EngineAddons.WallPotential.Radius = radius
settings.input.ams.Generator.Method = "CREST"
settings.input.ams.Output.KeepWorkDir = "Yes"
settings.input.ams.GeometryOptimization.MaxConvergenceTime = "High"
settings.input.ams.Generator.CREST.NCycles = 3 # at most 3 CREST cycles for this demo
settings.input.GFNFF = plams.Settings()
Run the conformers job¶
Now we can run the conformer generation job.
job = ConformersJob(molecule=mol, settings=settings)
job.run()
# ConformersJob.load_external("plams_workdir/conformers/conformers.rkf") # load from disk instead of running the job
[12.01|12:36:00] JOB conformers STARTED
[12.01|12:36:00] JOB conformers RUNNING
[12.01|12:41:35] JOB conformers FINISHED
[12.01|12:41:35] JOB conformers SUCCESSFUL
<scm.conformers.plams.interface.ConformersResults at 0x1731ef880>
rkf = job.results.rkfpath()
print(f"Conformers stored in {rkf}")
Conformers stored in /path/plams/examples/ConformersMultipleMolecules/plams_workdir/conformers/conformers.rkf
This job will run for approximately 15 minutes.
Results¶
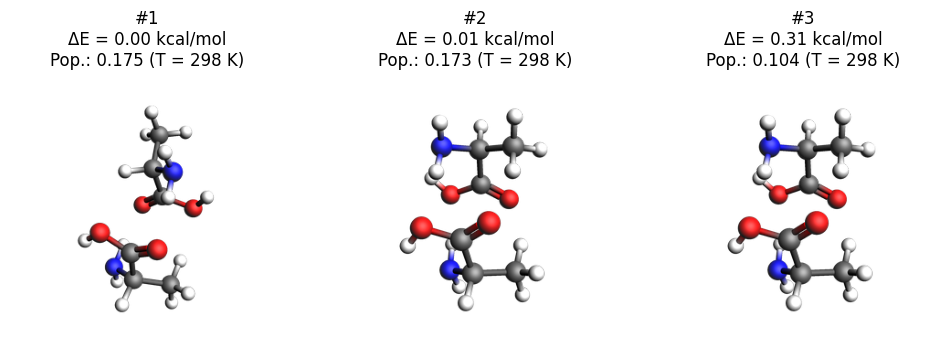
Here we plot the three lowest-energy conformers.
def plot_conformers(job: ConformersJob, indices=None, temperature=298, unit="kcal/mol", lowest=True):
molecules = job.results.get_conformers()
energies = job.results.get_relative_energies(unit)
populations = job.results.get_boltzmann_distribution(temperature)
if isinstance(indices, int):
N_plot = min(indices, len(energies))
if lowest:
indices = list(range(N_plot))
else:
indices = np.linspace(0, len(energies) - 1, N_plot, dtype=np.int32)
if indices is None:
indices = list(range(min(3, len(energies))))
fig, axes = plt.subplots(1, len(indices), figsize=(12, 4))
if len(indices) == 1:
axes = [axes]
for ax, i in zip(axes, indices):
mol = molecules[i]
E = energies[i]
population = populations[i]
if _has_view:
img = view(mol, width=300, height=300)
ax.imshow(img)
ax.axis("off")
ax.set_title(f"#{i+1}\nΔE = {E:.2f} kcal/mol\nPop.: {population:.3f} (T = {temperature} K)")
else:
view(mol, ax=ax)
ax.set_title(f"#{i+1}\nΔE = {E:.2f} kcal/mol\nPop.: {population:.3f} (T = {temperature} K)")
plot_conformers(job)
You can also open the conformers in AMSmovie to browse all conformers 1000+ conformers:
!amsmovie {rkf}
Finally in AMS2025+, you can also inspect the conformer data using the JobAnalysis tool.
try:
from scm.plams import JobAnalysis
ja = (
JobAnalysis(standard_fields=None)
.add_job(job)
.add_field(
"Id",
lambda j: list(range(1, len(j.results.get_conformers()) + 1)),
display_name="Conformer Id",
expansion_depth=1,
)
.add_field(
"Energies",
lambda j: j.results.get_relative_energies("kcal/mol"),
display_name="E",
expansion_depth=1,
fmt=".2f",
)
.add_field(
"Populations",
lambda j: j.results.get_boltzmann_distribution(298),
display_name="P",
expansion_depth=1,
fmt=".3f",
)
)
# Pretty-print if running in a notebook
if "ipykernel" in sys.modules:
ja.display_table(max_rows=20)
else:
print(ja.to_table())
except ImportError:
pass
Conformer Id |
E |
P |
|---|---|---|
1 |
0.00 |
0.175 |
2 |
0.01 |
0.173 |
3 |
0.31 |
0.104 |
4 |
0.33 |
0.100 |
5 |
0.59 |
0.065 |
6 |
0.87 |
0.040 |
7 |
0.89 |
0.039 |
8 |
1.10 |
0.028 |
9 |
1.14 |
0.026 |
10 |
1.36 |
0.018 |
… |
… |
… |
1062 |
256.89 |
0.000 |
1063 |
306.67 |
0.000 |
1064 |
326.40 |
0.000 |
1065 |
369.67 |
0.000 |
1066 |
371.07 |
0.000 |
1067 |
415.00 |
0.000 |
1068 |
415.08 |
0.000 |
1069 |
470.42 |
0.000 |
1070 |
502.31 |
0.000 |
1071 |
666.28 |
0.000 |
Complete Python code¶
#!/usr/bin/env amspython
# coding: utf-8
# Lets see how two alanine molecules orient themselves using CREST conformer generation.
# To do this we will constrain the system in a spherical region using the `SphericalWall` constraint.
# We start by setting up a system of two alanine molecules in a relatively small space.
# ## Initial imports
import scm.plams as plams
import sys
from scm.conformers import ConformersJob
import numpy as np
import matplotlib.pyplot as plt
import os
try:
from scm.plams import view # view molecule using AMSview in a Jupyter Notebook in AMS2026+
_has_view = True
except ImportError:
from scm.plams import plot_molecule # plot molecule in a Jupyter Notebook in AMS2023+
_has_view = False
def view(molecule, ax=None, **kwargs):
plot_molecule(molecule, ax=ax)
# this line is not required in AMS2025+
plams.init()
# ## Single alanine molecule
smiles = "CC(N)C(=O)O"
alanine = plams.from_smiles(smiles)
view(alanine, height=300, width=300)
# ## Initial system: alanine dimer
# Pack two alanine molecules in a sphere with a density of 0.5 kg/L.
density = 0.5
mol = plams.packmol(alanine, n_molecules=2, density=density, sphere=True)
# Translate the molecule to be centered around the origin (needed for SphericalWall later):
mol.translate(-np.array(mol.get_center_of_mass()))
view(mol, direction="along_pca3")
# ## Calculation setup
# To determine the radius of the `SphericalWall` we measure the size of the initial dimer.
dists = plams.distance_array(mol, mol)
max_dist = np.max(dists)
diameter = 1.33 * max_dist
radius = diameter / 2
print(f"Largest distance between atoms: {max_dist:.3f} ang.")
print(f"Radius: {radius:.3f} ang.")
# Now we can set up the Crest conformer generation job, with the appropriate spherical wall constraining the molecules close together.
settings = plams.Settings()
settings.input.ams.EngineAddons.WallPotential.Enabled = "Yes"
settings.input.ams.EngineAddons.WallPotential.Radius = radius
settings.input.ams.Generator.Method = "CREST"
settings.input.ams.Output.KeepWorkDir = "Yes"
settings.input.ams.GeometryOptimization.MaxConvergenceTime = "High"
settings.input.ams.Generator.CREST.NCycles = 3 # at most 3 CREST cycles for this demo
settings.input.GFNFF = plams.Settings()
# ## Run the conformers job
# Now we can run the conformer generation job.
job = ConformersJob(molecule=mol, settings=settings)
job.run()
# ConformersJob.load_external("plams_workdir/conformers/conformers.rkf") # load from disk instead of running the job
rkf = job.results.rkfpath()
print(f"Conformers stored in {rkf}")
# This job will run for approximately 15 minutes.
# ## Results
# Here we plot the three lowest-energy conformers.
def plot_conformers(job: ConformersJob, indices=None, temperature=298, unit="kcal/mol", lowest=True):
molecules = job.results.get_conformers()
energies = job.results.get_relative_energies(unit)
populations = job.results.get_boltzmann_distribution(temperature)
if isinstance(indices, int):
N_plot = min(indices, len(energies))
if lowest:
indices = list(range(N_plot))
else:
indices = np.linspace(0, len(energies) - 1, N_plot, dtype=np.int32)
if indices is None:
indices = list(range(min(3, len(energies))))
fig, axes = plt.subplots(1, len(indices), figsize=(12, 4))
if len(indices) == 1:
axes = [axes]
for ax, i in zip(axes, indices):
mol = molecules[i]
E = energies[i]
population = populations[i]
if _has_view:
img = view(mol, width=300, height=300)
ax.imshow(img)
ax.axis("off")
ax.set_title(f"#{i+1}\nΔE = {E:.2f} kcal/mol\nPop.: {population:.3f} (T = {temperature} K)")
else:
view(mol, ax=ax)
ax.set_title(f"#{i+1}\nΔE = {E:.2f} kcal/mol\nPop.: {population:.3f} (T = {temperature} K)")
plot_conformers(job)
# You can also open the conformers in AMSmovie to browse all conformers 1000+ conformers:
# Finally in AMS2025+, you can also inspect the conformer data using the JobAnalysis tool.
try:
from scm.plams import JobAnalysis
ja = (
JobAnalysis(standard_fields=None)
.add_job(job)
.add_field(
"Id",
lambda j: list(range(1, len(j.results.get_conformers()) + 1)),
display_name="Conformer Id",
expansion_depth=1,
)
.add_field(
"Energies",
lambda j: j.results.get_relative_energies("kcal/mol"),
display_name="E",
expansion_depth=1,
fmt=".2f",
)
.add_field(
"Populations",
lambda j: j.results.get_boltzmann_distribution(298),
display_name="P",
expansion_depth=1,
fmt=".3f",
)
)
# Pretty-print if running in a notebook
if "ipykernel" in sys.modules:
ja.display_table(max_rows=20)
else:
print(ja.to_table())
except ImportError:
pass