General¶
Introduction¶
The DFTB engine implements density functional based tight-binding methods, which can be viewed as computationally efficient approximations to density functional theory (DFT). As such it is a good engine for inexpensive calculations that still include quantum effects. DFTB is a computational engine that runs through the AMS driver. It can be used directly from the command line, from Python, and through our graphical interface.
What’s new in DFTB?¶
New in DFTB2025.1¶
Dispersion correction uses updated libraries for D3 (s-dftd3 1.2.1 https://github.com/dftd3/simple-dftd3) and D4 (dftd4 3.7.0 https://github.com/dftd4/dftd4)
QM/FQ ground-state gradients and geometry optimizations are now available with DFTB.
For EffectiveMass the position of the HOMO and LUMO is determined along the high-symmetry path. To get the pre-2025 behavior, set useBandStructureInfoFromPath=No.
Changed default:
Before AMS2025 the band structure (if requested) was not calculated if only 1 k-point was used. In AMS2025 this exception has been removed. If you do not need it, use BandStructure%Enabled=No.
New in DFTB2024.1¶
Default DOS is now divided by DeltaE, which scales the overall DOS and PDOS so that they have the standard unit (1/(energy*volume)).
New in DFTB2023.1¶
Improved SCC convergence with the MultiStepper.
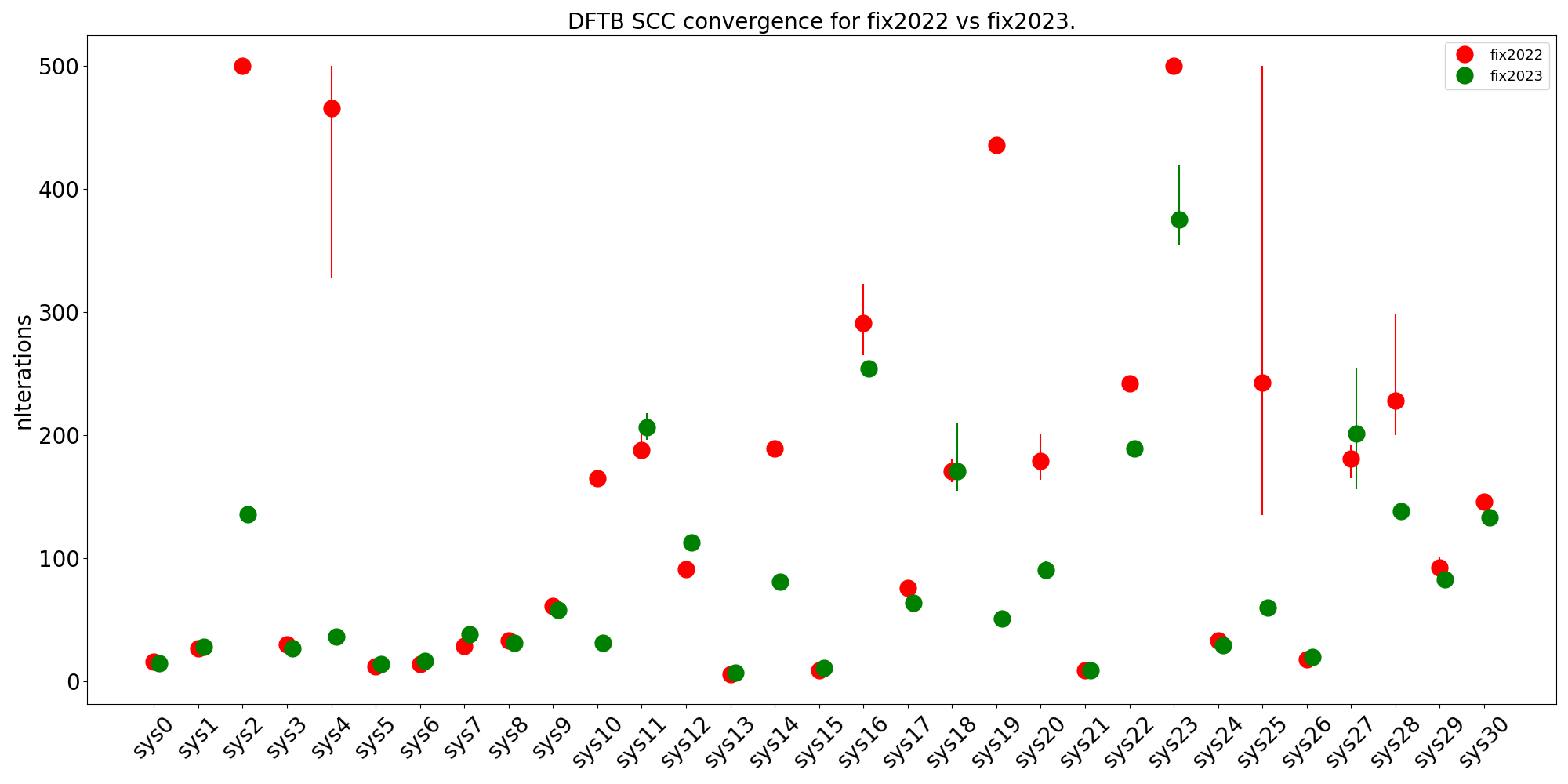
Fig. 1 Comparison of the number of SCC cycles needed. For easy systems there is not much difference, for more difficult systems, however, the fix2023 (green) is an improvement over the fix2022 (red). As there can be some randomness in the number of iterations the calculations are repeated five times (using a different number of cores), the dot is the average number of cycles used, and the vertical lines show the spread in the number of iterations (if any). The maximum number of iterations was set to 500.¶
New in DFTB2022.1¶
Visualization of orbitals in AMSview now also works for calculations with (most) DFTB.org parameter sets.
New in DFTB2021.1¶
The D4 dispersion correction has been added. It can be used with the Slater-Koster based model Hamiltonians and the DFTB.org parameter sets.
New in DFTB2020¶
Calculations with the GFN1-xTB model and many k-points are significantly faster.
The default model has been changed from SCC-DFTB to GFN1-xTB, as the latter supports all elements.
Various new applications in the AMS driver.
New in DFTB2019.3¶
The internals of the DFTB engine have been restructured, making it faster, more scalable and more accurate for periodic systems, while at the same time enabling previously locked combinations of features:
The default for the accuracy of k-space integration has been changed: DFTB used to sample only the Γ-point by default. As of this release the default k-points depend on the system size, using the same logic as in BAND. See the page on k-space integration in the BAND manual.
Calculations with k-space integration are generally faster and scale much better on parallel machines.
The GFN1-xTB model can now be used together with k-space integration.
Unrestricted calculations can now also be performed in conjunction with k-space integration.
The orbital dependent (l-dependent) SCC cycle is now compatible with k-space integration.
The stress tensor is now calculated analytically, making its calculation faster and the result more accurate.
An implicit solvation model (GBSA: Generalized Born (GB) model augmented with the solvent accessible surface area (SA) term) has been added to DFTB, allowing simulations of molecules in solution.
Various new applications in the AMS driver.
New in DFTB2019.1¶
Grimme’s GFN1-xTB has been added as a new model Hamiltonian. It supports molecular as well as periodic calculations for systems including elements up to Radon. Visualization of the results (e.g. molecular orbitals) in AMSview is also supported.
Various new applications in the AMS driver.
More robust and easier to set up k-space integration.
More robust SCC convergence:
Adaptive mixing: The charge mixing parameter is automatically decreased if the energy increases during the SCC cycle.
The default electronic temperature has been increased to 300K, making SCC convergence more robust for systems with small HOMO-LUMO gaps.
New in DFTB2018¶
New features¶
Elastic tensor and related properties (e.g. Bulk modulus) (via AMS driver)
Linear transit and PES scan (via AMS driver)
Geometry optimization under pressure (via AMS driver)
…
AMS: a new driver program¶
Important
In the 2018 release of the Amsterdam Modeling Suite we introduced a new driver program call AMS. We recommend you first read the General section of the AMS Manual
If you use DFTB exclusively via the Graphical User Interface (GUI), this change should not create any issues. If, on the other hand, you create input files by hand (or you use DFTB via PLAMS), then you should be aware that shell scripts for DFTB2017 and previous versions are not compatible with DFTB2019 and have to be adjusted to the new setup.
The example below shows how a shell script for DFTB2017 is converted to DFTB2019.
DFTB2017 shell script (obsolete):
#!/bin/sh
# This is a shell script for DFTB2017 which will not work for DFTB2019
$AMSBIN/dftb << EOF
Task
RunType GO
End
System
Atoms
H 0.0 0.0 0.0
H 0.9 0.0 0.0
End
End
DFTB
ResourcesDir Dresden
End
Geometry
iterations 100
End
EOF
DFTB2019 shell script:
#!/bin/sh
# This is a shell script for DFTB2019
# The executable '$AMSBIN/dftb' is no longer present.
# You should use '$AMSBIN/ams' instead.
$AMSBIN/ams << EOF
# Input options for the AMS driver:
System
Atoms
H 0.0 0.0 0.0
H 0.9 0.0 0.0
End
End
Task GeometryOptimization
GeometryOptimization
MaxIterations 100
End
# The input options for DFTB, which are described in this manual,
# should be specified in the 'Engine DFTB' block:
Engine DFTB
ResourcesDir Dresden
EndEngine
EOF