Building Molecules¶
In the previous tutorial you have learned how to construct a molecule by building it out of atoms. For bigger molecules this process can be quite time-consuming. Therefore, AMSinput provides various tools to help you with building molecules.
This tutorial will cover easy ways to add molecules or solvents:
The quickest is to search (
 ) for a molecule inside AMSinput, using available structures from the built-in database
) for a molecule inside AMSinput, using available structures from the built-in databaseWhen searching for molecules on the Internet, in open-source datasets or with chemical suppliers, you can directly load provided xyz files or SMILES strings
We show how to use the structure tool in AMSinput to build larger molecules:
Building a small peptide chain
Building metallo-organic complexes
Creating your own structures library
Finally, you can use the crystal tools to:
Cut a nanoparticle from a crystal
Create carbon nanotubes
Start AMSinput¶
We start by creating a new folder where we will save our molecules:
Search for ethanol¶
For common molecules, the easiest way to get the coordinates is to search for them in AMSinput.
in the panel bar)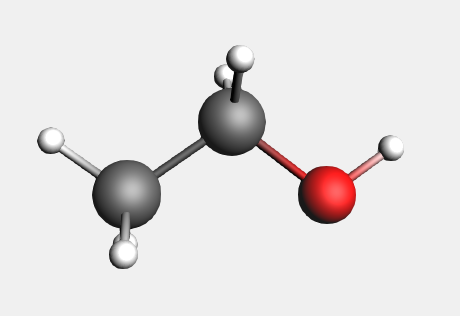
Your ethanol is ready. The (ADF) in the search results means that the molecule has already been optimized by ADF. (Unless states otherwise, these optimizations were done with the BP86 XC potential, TZP basis set and small core.)
Import XYZ for ethanol¶
AMSinput allows importing coordinates from various different file types, including the .ams files obtained when running jobs with AMS.
For files obtained from online sources, the xyz format is the most common.
AMSinput can also be used to generate these xyz files:
Your xyz file should now be visible in the AMSjobs browser:
AMSinput will start and automatically import the .xyz file. To import a molecule when AMSinput is already open (for instance, if you want to add solvent molecules), you can use the File → Import Coordinates… command:
Tip
It is also possible to copy/paste data directly into AMSinput. This works for many formats, including xyz, SMILES or InChI strings.
The coordinates for ethanol are:
C 0.01247000 0.02254000 1.08262000
C -0.00894000 -0.01624000 -0.43421000
H -0.49334000 0.93505000 1.44716000
H 1.05522000 0.04512000 1.44808000
H -0.64695000 -1.12346000 2.54219000
H 0.50112000 -0.91640000 -0.80440000
H 0.49999000 0.86726000 -0.84481000
H -1.04310000 -0.02739000 -0.80544000
O -0.66442000 -1.15471000 1.56909000
Import SMILES string¶
AMSinput can also interpret SMILES strings. In case of ethanol:
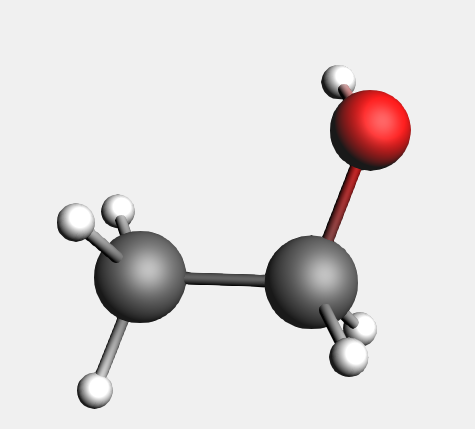
Because SMILES strings do not contain information on the 3D structure, the coordinates that we obtain are different from the optimized structure from ADF.
It is typically recommended to first pre-optimize ( ) the structure with UFF when loading from SMILES. This results in more reasonable structures and helps speed up subsequent simulations done using e.g. ADF or BAND.
) the structure with UFF when loading from SMILES. This results in more reasonable structures and helps speed up subsequent simulations done using e.g. ADF or BAND.
Build ethanol using the structure tool¶
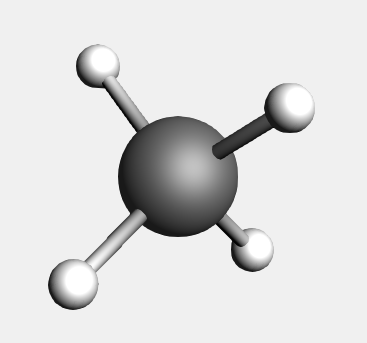
As a demonstration on how to use the structure tool  , we start by building a methane molecule:
, we start by building a methane molecule:
The next step is to add a methyl group, using the structure tool :
→ Alkyl Chains → Methyl structureNotice that the button of the structures menu is now glowing, indicating that the structure tool is active.
You will see that the hydrogen is replaced by a methyl group.
Note that:
The methyl is orientated along the newly formed C-C bond and the new hydrogens point away from the existing ones.
The carbon on the newly-placed methyl group was designated as the ‘replacing’ atom. This carbon atom has been placed at the location of the substituted hydrogen atom. All structures in the structure tool have a predefined ‘replacing’ atom.
The background glow from the ‘Structures’ tool has moved to the ‘Pointer’ tool button; the ‘Pointer’ tool is active again.
To create ethanol, we need to add a hydroxyl group:
→ Ligands → OH structure
Again, the hydrogen is replaced by the structure. In this case, the oxygen replaces the selected atom. The O-H bond is aligned along the C-O bond and points away from the rest of the molecule. This shows you the very general way in which the structures will align according to the bonds in the original molecule and those in the structure. In this case, the hydroxyl group is not automatically oriented as it normally would be in an ethanol molecule.
Add solvent molecules using the structure tool¶
The structure tool includes several commonly-used solvent molecules, allowing you to easily add solvents to your system. Ethanol is also included in this list:
→ Solvents → Ethanol structureWe can select the new molecule to orient it to a different position:
Building a peptide chain using the structure tool¶
Larger biomolecules or macromolecules can be assembled using the basic building blocks in the structure tool. We will look at a small peptide chain as an example.
→ Amino Acids → AA Backbone structure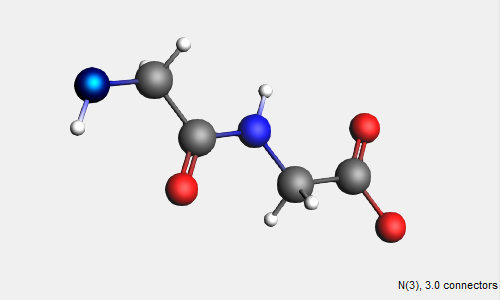
The AA backbone structure contains 2 subunits of a basic peptide chain. We can extend the peptide backbone by adding additional subunits.
Tip
Press the space bar to reuse the previous structure tool
→ Amino Acids → AA Backbone structure (or just press the space bar)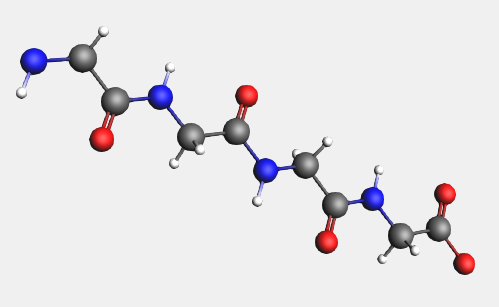
In a similar fashion, you can replace the hydrogens on the backbone by amino acid side groups of your choice. These can be found in the → Amino Acid → AA Side Groups sub-menu.
Metal complexes and ligands¶
In the sub-menu ‘Metal Complexes’ you can find a set of predefined complexes corresponding to commonly encountered geometries. A set of ligands has also been provided, including typical monodentates, bidentates and polydentates. These can be used to quickly construct metallo-organic structures.
Predefined Metal Complex Geometries¶
→ Metal Complexes → ML6 Octahedral structure and place it in the drawing areaNotice that six dummy (“Xx”) atoms have been placed around the metal center in an octahedral fashion.
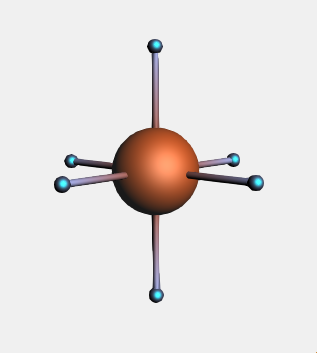
When replacing multiple atoms with the same side-group, instead of using the structure tool, we can access the ligands selection through the Atoms menu:
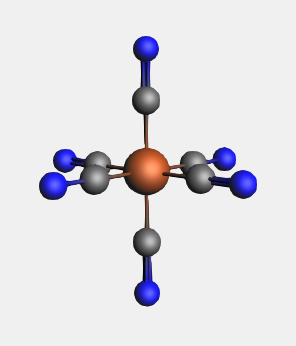
Notice that all the dummy atoms in the selection have been replaced by CN ligands.
Bidentate Ligands¶
In order to use the bidentate ligands, we must start with a bare metal center.
→ Ligands → Bidentates → Ethylenediamine structure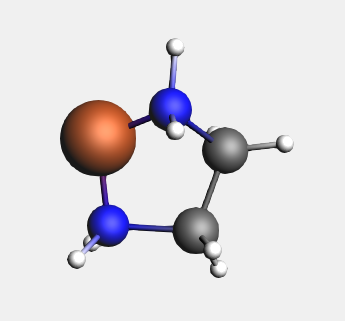
For the bidentate ligands, the Structure Tool attaches the ligand to the selected atom, instead of replacing it. Other multidentate ligands are defined in a similar fashion.
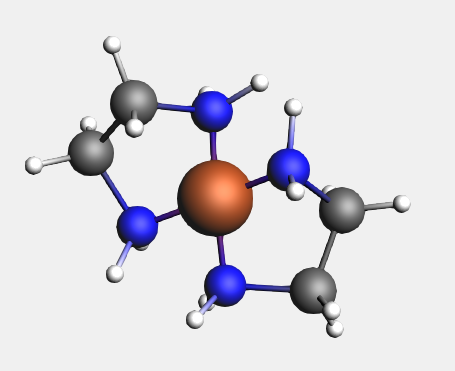
Notice that the second ligand appears opposite the existing one.
Modifying the Plane Angle¶
To change the relative orientation of two bidentate ligands, we can change the plane angle. The planes are defined by two sets of three atoms, the central one being present in both sets. In this case this will, of course, be the metal atom.
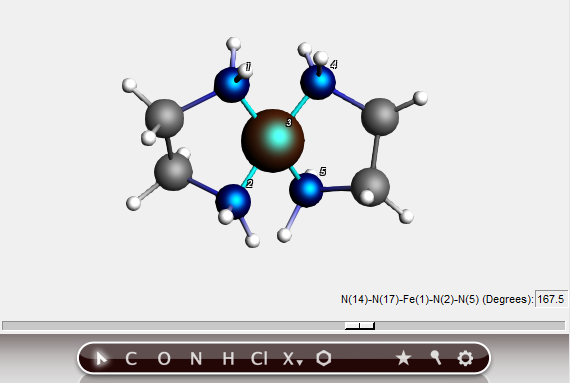
In this way, you can easily change the environment around the metal from square planar to tetrahedral. This feature works as long as you choose the atoms in the right order, and if the defined planes can freely rotate relative to each other.
Your own structures library¶
You can make your own structure library for use with the Structure Tool. This allows you to easily access frequently-used building blocks that are relevant to your research.
By default, user-defined structures will be stored in the .scm_gui/Structures directory.
Defining your structures¶
To be able to actually use the structures as described earlier, it is necessary to define one of the atoms as having xyz-coordinates (0,0,0). This atom will be designated as the ‘replacing’ atom. By using the ‘Save As Structure’ command, this step will be done for you.
the structure → Save As Structure … command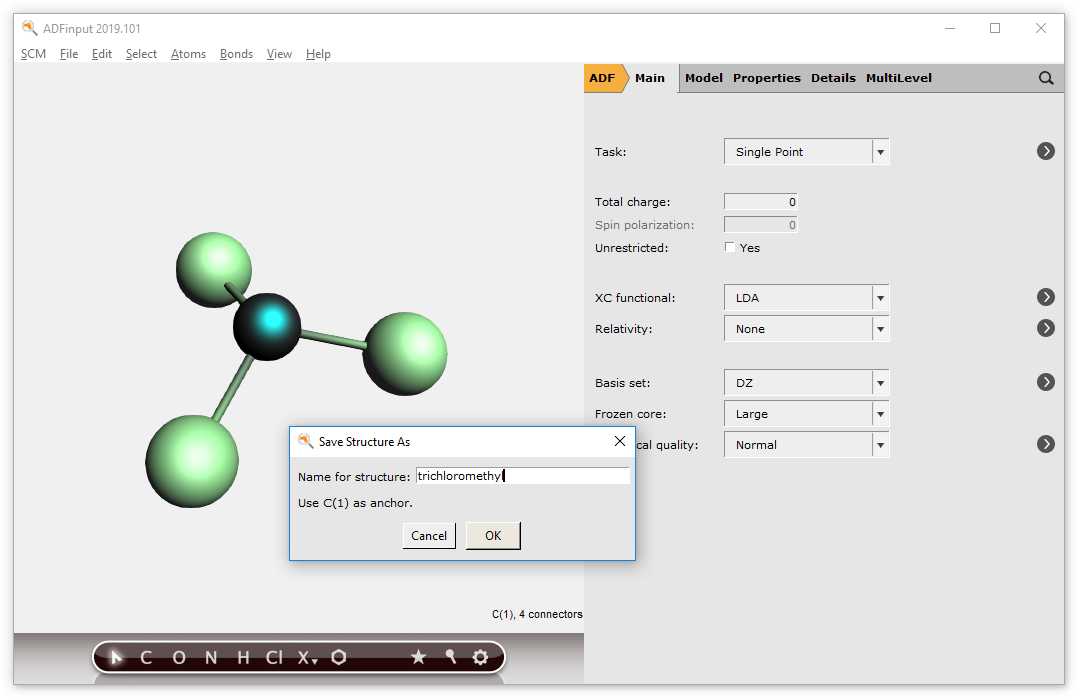
The new structure will appear in the Structures menu and can be used immediately.
Using dummy atoms¶
Dummy (“Xx”) atoms are treated differently when used in structures. A dummy atom will not replace an existing atom when it is defined as the ‘replacing atom’. Instead, the double-clicked atom will remain and will accept the bonds that the dummy atom had in the structure. (As with the bidentates shown earlier.)
→ Save As Structure … command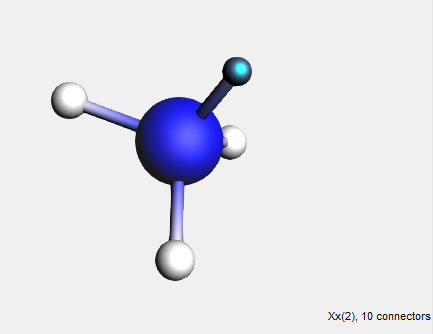
Because a dummy atom was used, the NH3 group has been attached to the hydrogen atom instead of replacing it. This way, dummy atoms can be used to conveniently add complex building blocks such as multidentate ligands or monomer units.
If you want to clean up your structures, you can use the → Manage Structures… command. If you use it, AMSjobs will open and show the contents of your Structures directory. As the structures are just (simplified) .ams files, you can open them using AMSinput for viewing or editing. Using AMSjobs, you can rename or delete the files. The Structure Tool will update automatically.
A sphere of Cu atoms, cut out of the crystal¶
When constructing single crystals or nanoparticles, it is often easiest to start from a crystalline bulk material. We will showcase this procedure here for a copper particle.
We start by making a Cu crystal. We will use a super cell to obtain a large number of Cu atoms.
To build the crystal, we need to use the Periodic tools. These will work only for engines supporting periodicity (e.g BAND).
) for ‘copper’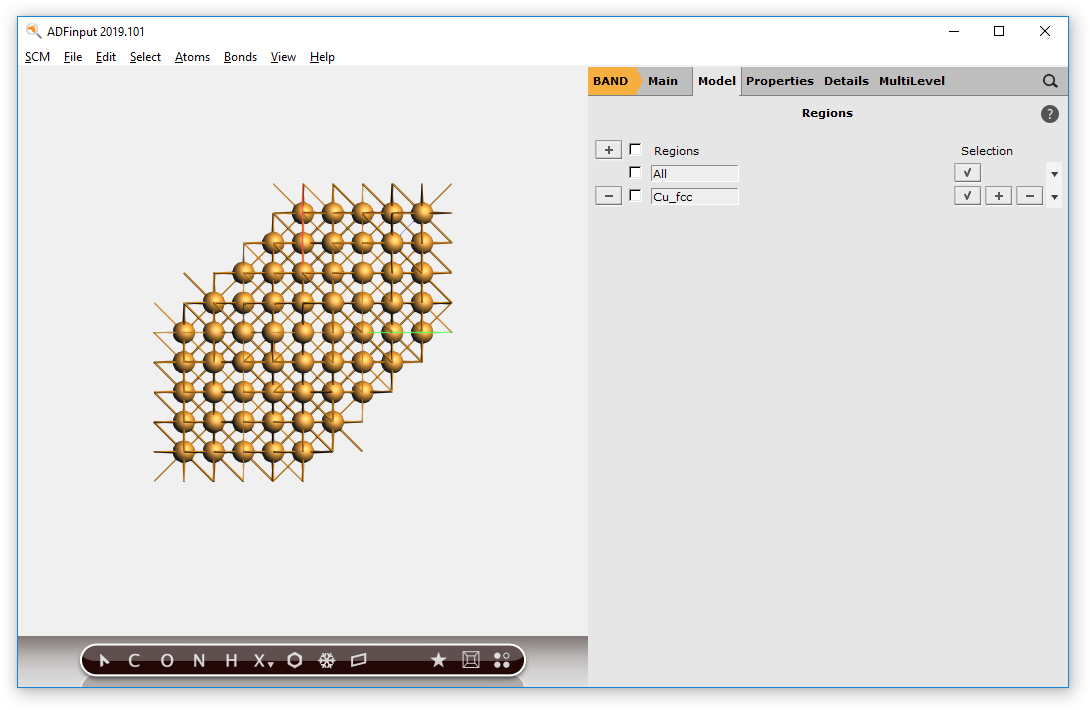
Now we have a block of Cu, with explicit Cu atoms. Next we will center this block, and select a sphere of atoms around the origin.
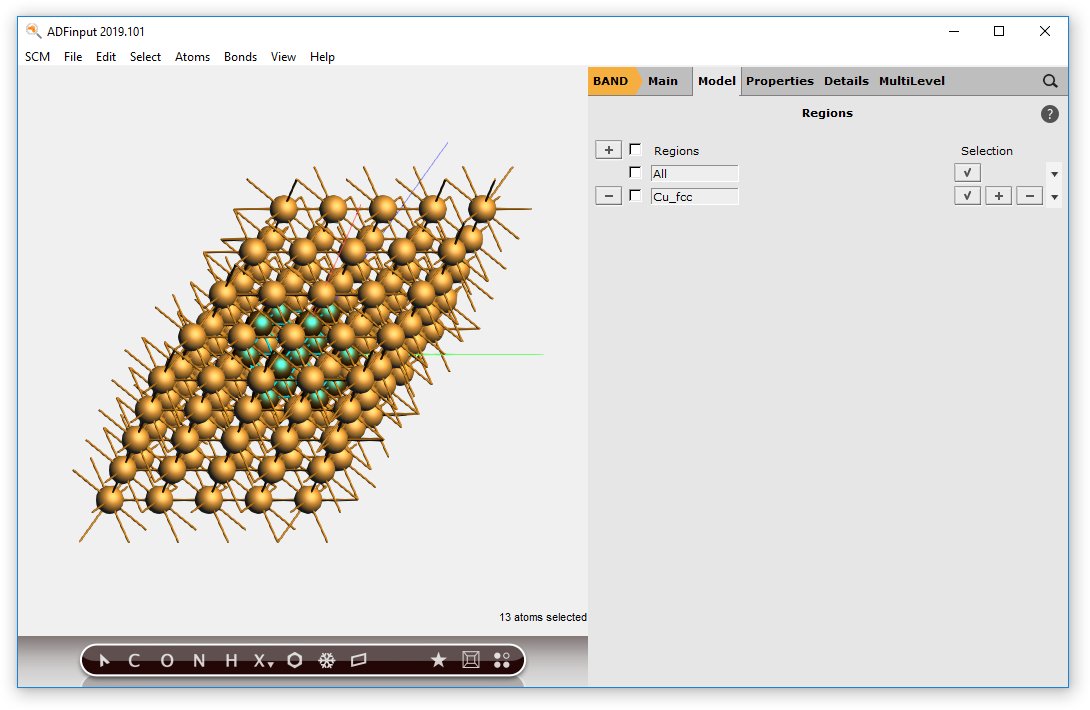
You should now have a (very small) sphere consisting of Cu atoms in the molecular ADF program:
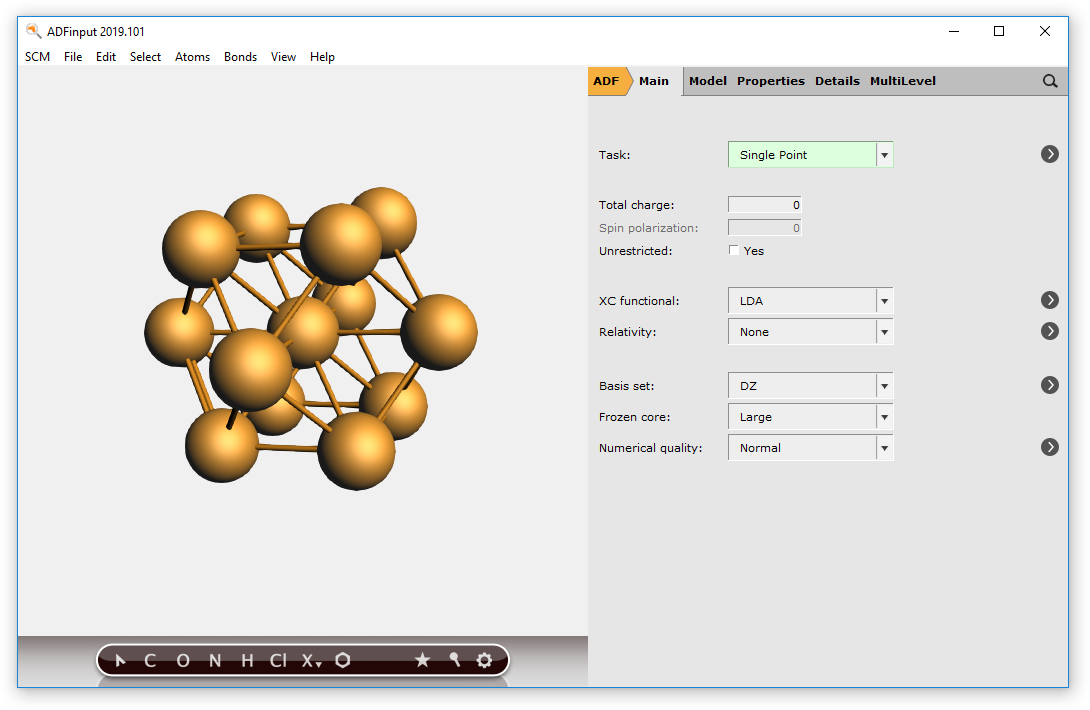
To make bigger spheres, simply choose a bigger super cell and use a larger radius when selecting atoms.
Tip
For building mono-metallic Wulff particles, one can also use the dedicated nanoparticle builder.
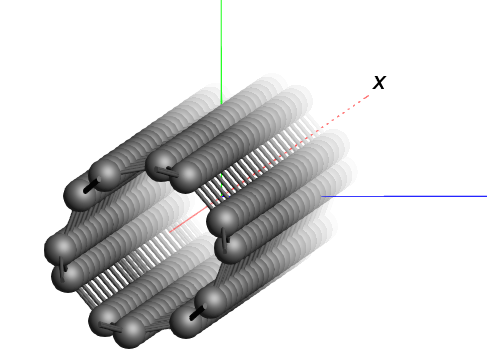
Working with nanotubes¶
A carbon nanotube segment is included in the AMSinput database ().
Nanotubes with alternate structures or elemental compositions can be obtained through online databases, or by using the free TubeGen tool.
Because nanotubes are periodic structures, they are typically provided as CIF files:
data_nanotube
_audit_creation_method '(3,3) Nanotube -- TubeGen 3.3, J T Frey, University of Delaware'
_cell_length_a 7.4762
_cell_length_b 7.4762
_cell_length_c 2.4643
_cell_angle_alpha 90.00
_cell_angle_beta 90.00
_cell_angle_gamma 120.00
_symmetry_space_group_name_H-M 'P 1'
_symmetry_Int_Tables_number 1
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
C 0.7762 0.5000 0.0000
C 0.8138 0.7061 0.0000
C 0.7762 0.7762 0.5000
C 0.6077 0.8138 0.5000
C 0.5000 0.7762 0.0000
C 0.2939 0.6077 0.0000
C 0.2238 0.5000 0.5000
C 0.1862 0.2939 0.5000
C 0.2238 0.2238 0.0000
C 0.3923 0.1862 0.0000
C 0.5000 0.2238 0.5000
C 0.7061 0.3923 0.5000
Now we want to get this structure into AMSinput:
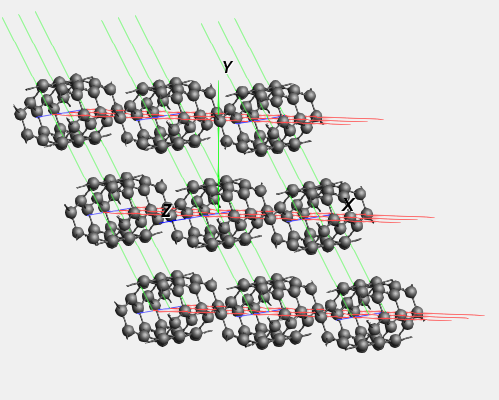
We see a piece of nanotube, which is repeated in all directions. In the periodic view, 9 nanotubes are visible side-by-side.
In order to obtain a model of a single nanotube, we can set the periodicity to be one-dimensional in AMSinput. 1D systems in AMS always assume that the structure is oriented along the X-axis. However, the structure that we imported here has the nanotubes oriented along the Z-axis instead. To change our nanotube structure into a single tube, we therefore need to rotate it first.
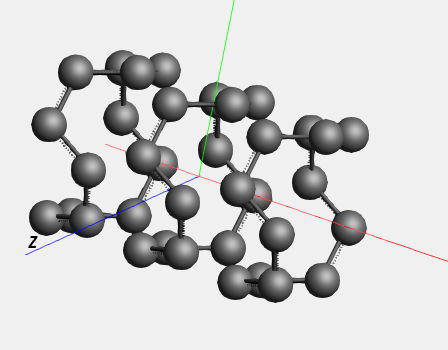
The ‘Rotate 90’ command not only rotated the coordinates of the atoms, but also the lattice vectors.
We now have a small piece of nanotube. (Since the periodic view already includes adjacent cells, the repeat unit is actually quite small.) If you want to build a larger model, this is easily achieved using the super cell method:
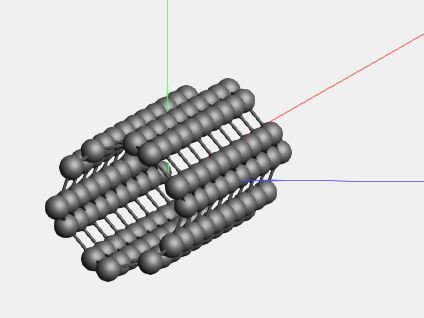
Switch to some non-periodic code (like ADF) if you wish to treat this piece of nanotube without infinite symmetry.
If you have a large system you can sometimes get a better view by introducing Fog.