Building Liquids and Gases¶
Introduction¶
This tutorial uses the new Builder introduced in AMS2025.
This tutorial shows how to build liquids and gases, and solid-liquid and solid-gas interfaces.
The packing is done using the Packmol program, that is included with AMS. The interface to Packmol is integrated into AMSinput under Builders → Packed molecules.
See also
Documentation for Builder
The two most important starting modes of the Builder¶
The Builder can either create an entirely new system or pack around atoms in an existing system, depending on the current system when the Builder is started.
Packing in an empty box: The Builder is started for an empty system. The Builder will then pack the molecules and modify the lattice vectors of the system.
Packing inside the crystal: The Builder is started for a non-empty 3D-periodic system. The Builder will then pack molecules around the existing system, into a space defined by the lattice vectors. The lattice vectors will not change.
For more details, see the Builder documentation.
The Builder has flexible input¶
The Builder has flexible input. This means that you specify only some of the input options, and appropriate or approximate values for the remaining input options will be automatically determined.
Liquid water¶
Start the Builder¶
 →
→ 
This starts the builder in the Packing in empty box mode.
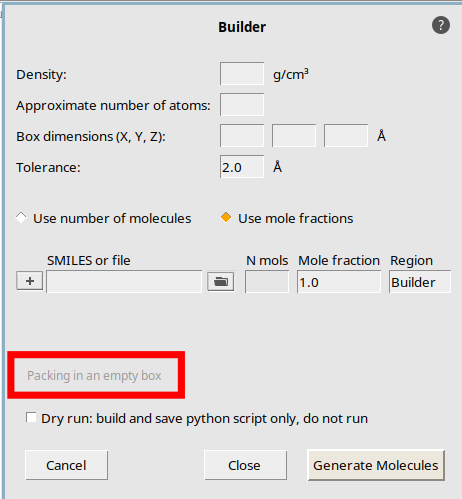
Tip
Activate the engine that you want to use for your calculation before
starting the Builder, for example by switching from →
.
Why? The Builder is most commonly used to build a 3D-periodic system. However, the ADF engine (default in AMSinput) cannot handle 3D-periodic systems. If the ADF engine is active when you start the builder, then the lattice vectors will be removed when you close the builder, since ADF cannot handle them.
Density, number of atoms, SMILES¶
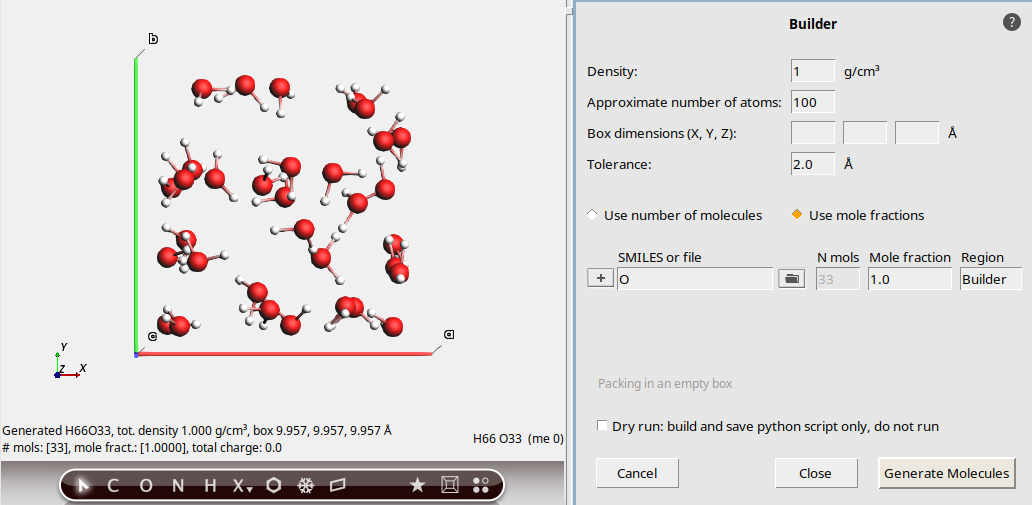
1.0 g/cm3100O (capital letter O, which is the SMILES code for water)This updates the molecule view on the left hand side.
Always double-check results¶
You should always double-check the results from the Builder.
No box dimensions were specified, so a cubic system is created with a volume that matches the specified density.
We specified 100 atoms. The Builder translated this into
33water molecules, giving a total of 99 atoms. When specifying the number of atoms, it is always approximate.
In the bottom-left corner, some information about the new system is printed, for example the total density and lattice vectors.
Rerun with different settings¶
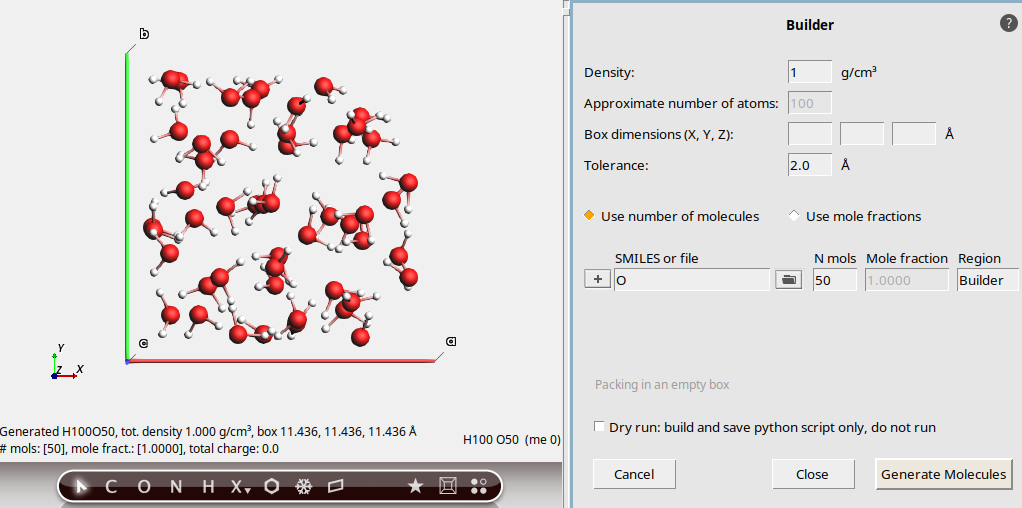
Clicking the Generate molecules button a second time will discard the previous run and rerun with the new settings.
50This updates the molecule view on the left. There are now 50 water molecules instead.
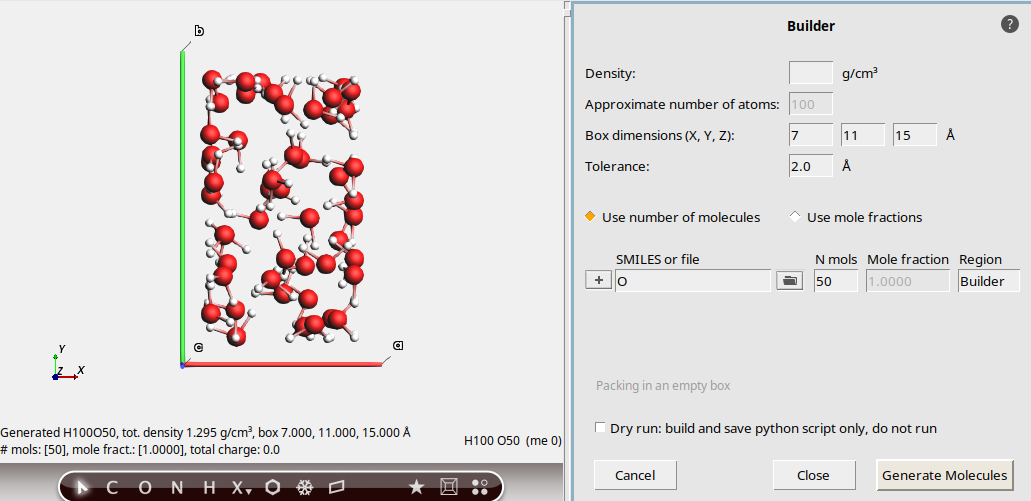
Pack in a custom box¶
7, 11, 15 ÅThis packs 50 water molecules into a box of the specified dimensions.
You can see the density at the bottom left.
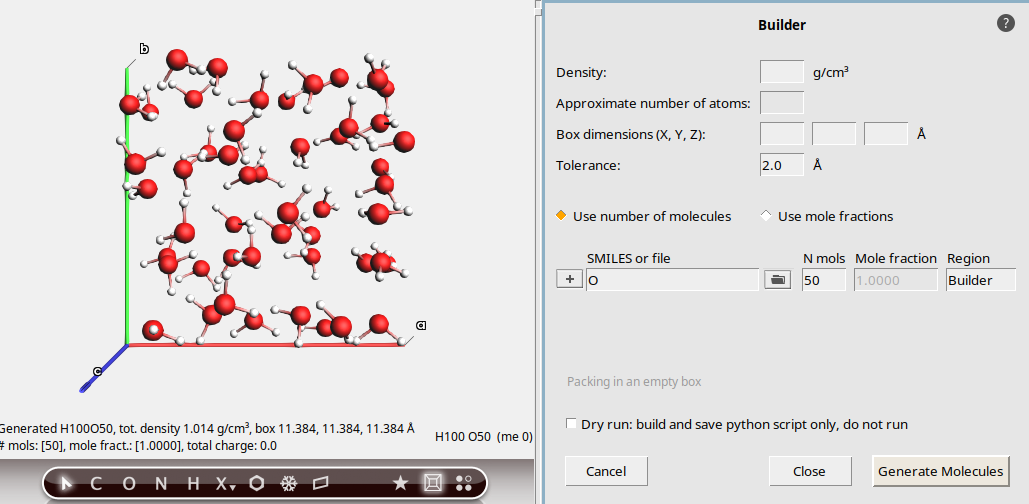
Guess the density¶
The Builder can guess a meaningful density. However, this density is often quite inaccurate. If you use this feature, always make sure to equilibrate the density with for example an NPT molecular dynamics simulation at a given pressure and temperature.
50This generates a cubic box of 50 water molecules with density 1.01 g/cm3.
There are also more possible combinations of input options. Feel free to experiment with different possibilities.
Close the builder¶
This brings you back to the main AMSinput window. The periodicity of the system is set to Bulk and the lattice vectors match those from the builder:
Mixture of water and acetonitrile¶
→ This starts the builder in the Packing in empty box mode.
Dilute solution¶
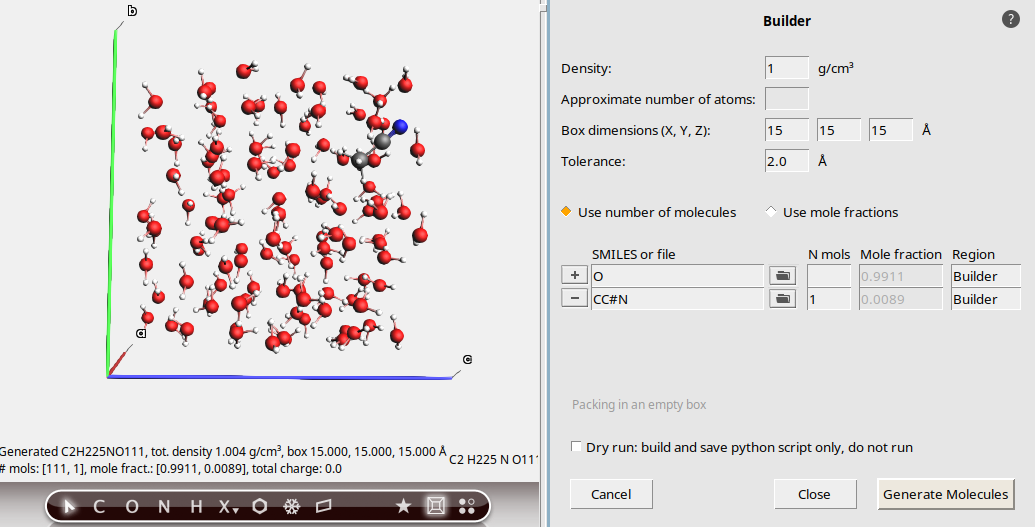
New in AMS2026: Fill up a box with a given number of solute molecules, and the rest with an automatically determined number of solvent molecules.
1.0 g/cm315, 15, 15 Å.O (SMILES for water) button to add a new molecule
button to add a new moleculeCC#N (SMILES for acetonitrile)1This fills up the box with an automatically determined number of water (solvent) molecules around the solute, to match the density and box dimensions.
Mole fractions, AMS structure library¶
To build a water-acetointrile mixture, we could use the SMILES codes (O
and CC#N) as before. However, here we demonstrate how to import
structures from the AMS structure library.
0.8 g/cm3300water and click on the first match in the popup. The text will change to SCM/Water.xyz button to add a new moleculeacetonitrile and click on the first match in the popup. The text will change to SCM/Acetonitrile.xyzWith these settings, the Builder will attempt to pack water and acetonitrile in a 1:2 ratio.
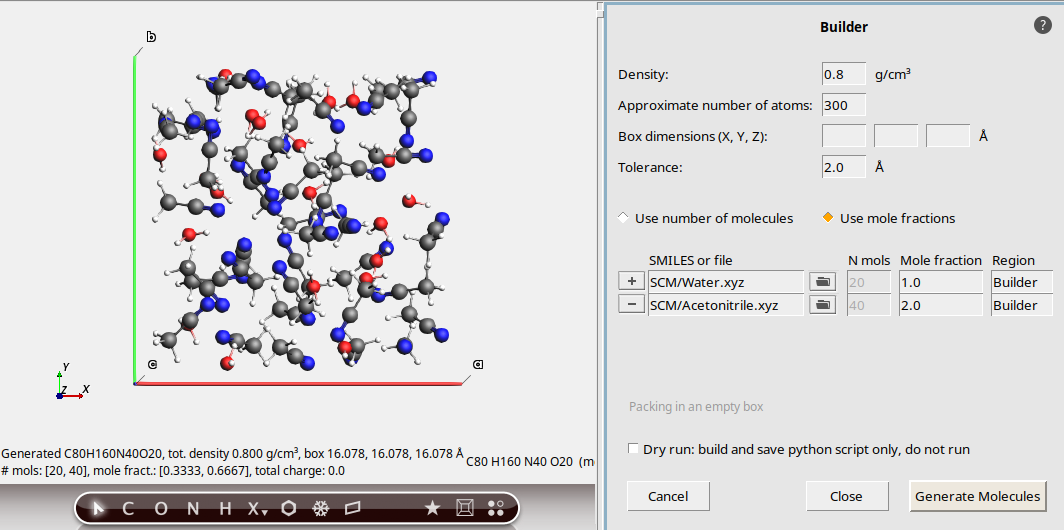
The resulting system has 20 water molecules and 40 acetonitrile molecules, the
density is as specified, and the number of atoms happens to be exactly 300.
The mole fractions are always approximate¶
Just like the number of atoms are approximate, so are the mole fractions:
200This replaces the system, but the Mole fraction column is now highlighted in red. This indicates that the mole fractions in the final system do not exactly match the input mole fractions.
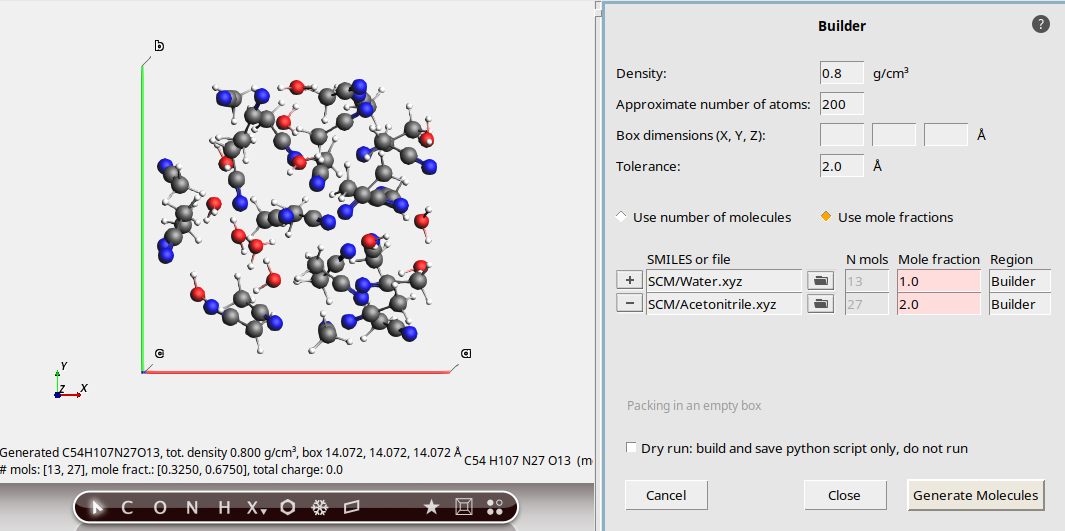
In the greyed-out N mols column, you can see that the Builder packed 13 water molecules and 27 acetonitrile molecules. So the ratio is not exactly 1:2.
The Builder deviated from the specified mole fractions to more closely match the requested number of atoms, which was set to 200.
This shows that the system has 201 atoms.
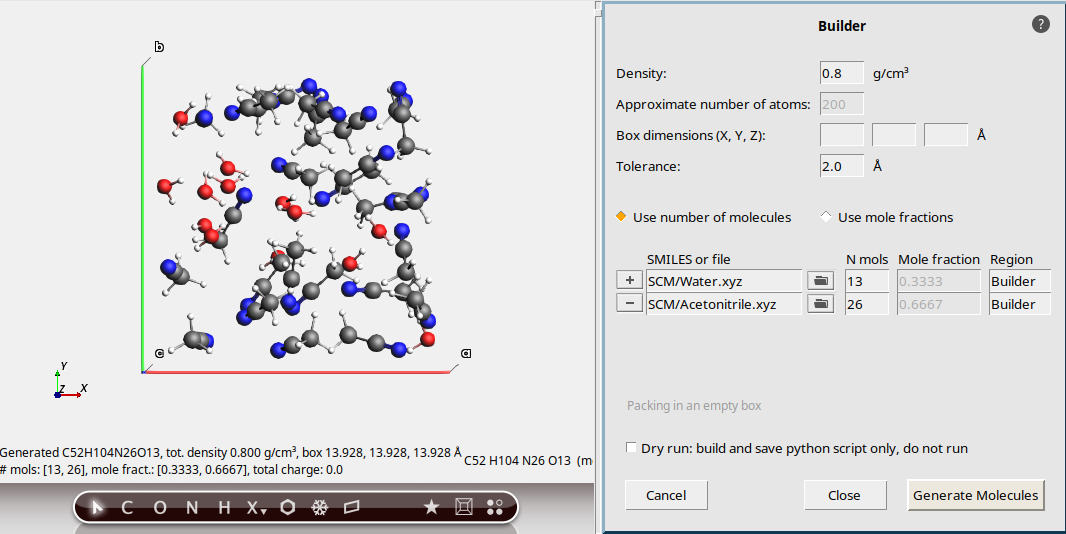
Manually modify the number of molecules to get correct mole fractions¶
If you want to ensure that the ratio of water:acetonitrile is exactly 1:2, you must specify the number of molecules. In this case, we just need to subtract 1 from the number of acetonitrile:
26This gives a 1:2 water:acetonitrile mixture.
Adjust the tolerance for tighter packing¶
It is usually quite easy for the Builder pack small molecules like water and acetonitrile.
For larger molecules, the packing may either fail, or take a very long time. There are several possible solutions to this:
Decrease the density
Provide larger box dimensions
Decrease the tolerance
Let’s illustrate the effect of the tolerance. We will keep the water-acetonitrile example, but try to pack at higher density:
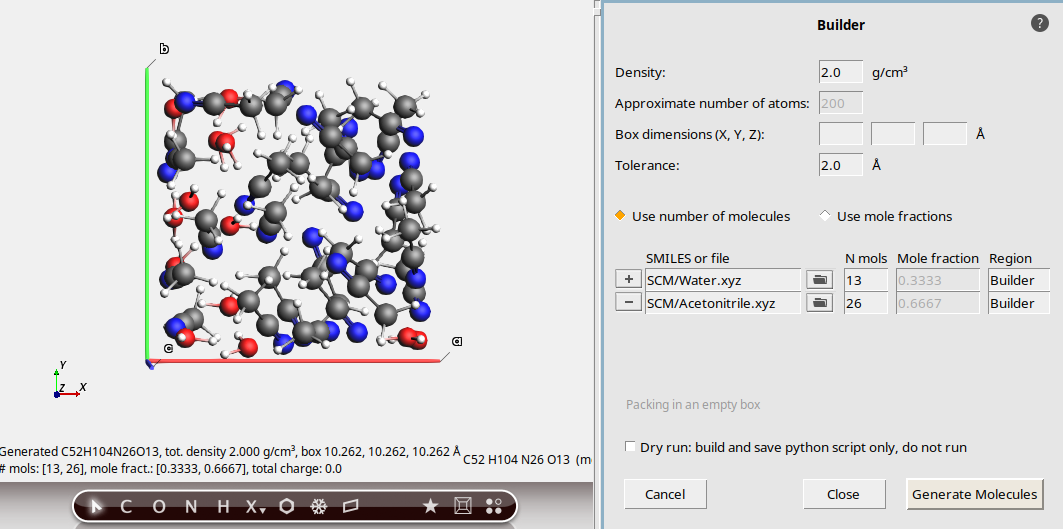
Note that it takes quite some time for the packing to finish.
This is an unrealistically high density for this mixture. We can make the job of the Builder easier by decreasing the Tolerance. This will allow the Builder to use smaller intermolecular distances.
Now the packing was successful much faster.
Warning
If you specify very high densities, the Builder may cause molecules to overlap. Always check that the density is reasonable.
Pack around an existing system¶
If you have a 3D-periodic system with at least 1 atom in it, the Builder will start in the Packing inside crystal mode:
→ System
Atoms
Cu 7.23252386 0.0 7.9157655282
Cu 5.95619612 0.7368881643 10.0
Cu 4.67986838 1.4737763257 12.0842344718
Cu 7.23252386 1.4737763257 12.0842344718
Cu 0.85088516 0.7368881643 10.0
Cu 2.1272129 0.0 7.9157655282
Cu 3.40354064 3.6844408157 12.0842344718
Cu 4.67986838 2.9475526543 10.0
Cu 5.95619612 2.21066449 7.9157655282
Cu 2.1272129 1.4737763257 12.0842344718
Cu 3.40354064 0.7368881643 10.0
Cu 4.67986838 0.0 7.9157655282
Cu 0.85088516 3.6844408157 12.0842344718
Cu 2.1272129 2.9475526543 10.0
Cu 3.40354064 2.21066449 7.9157655282
Cu 5.95619612 3.6844408157 12.0842344718
Cu 7.23252386 2.9475526543 10.0
Cu 0.85088516 2.21066449 7.9157655282
End
Lattice
7.65796644 0.0 0.0
0.0 4.42132898 0.0
0.0 0.0 20.0
End
End
This is a Cu(111) slab with a vacuum gap long the z direction. The unit cell is orthorhombic with vectors parallel to x, y, and z.
0.8 g/cm3CO (SMILES code for methanol)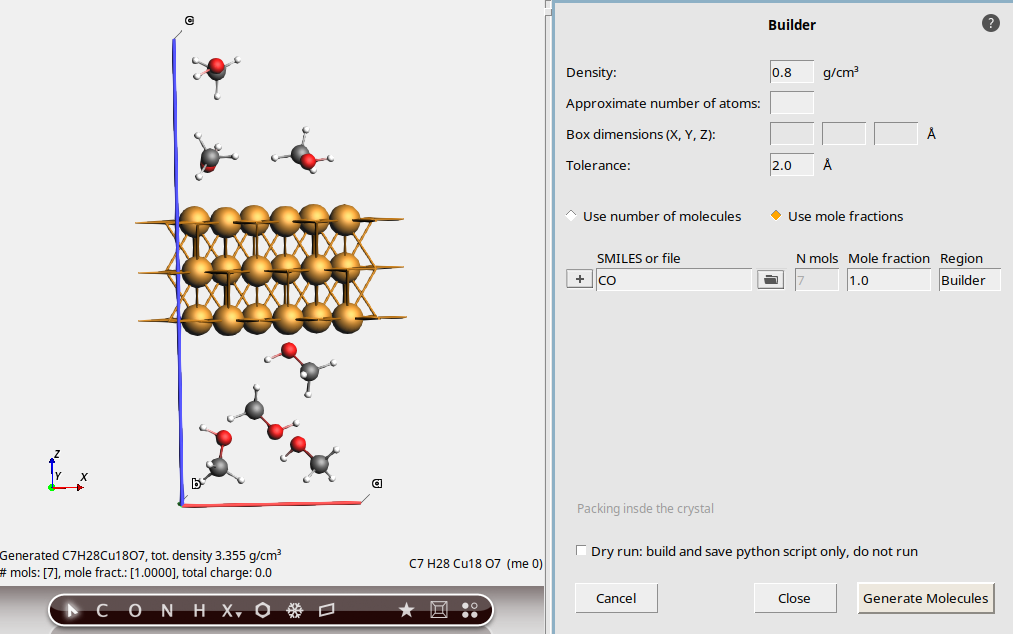
This packs 7 methanol molecules into the vacuum region. The density is approximately 0.8 g/cm3. An estimate is made for the available free volume to pack the molecules in. This can be quite inaccurate, especially if the original system is not a metal. Moreover, the estimate does not take any surface interactions between the surface and the methanol into account.
For better control of the number of water molecules, tick Use number of molecules and set N mols. As before, clicking Generate molecules multiple times will discard the last build and rerun with new settings.
7 (it should already be filled in)This adds 7 methanol molecules into the vacuum region.
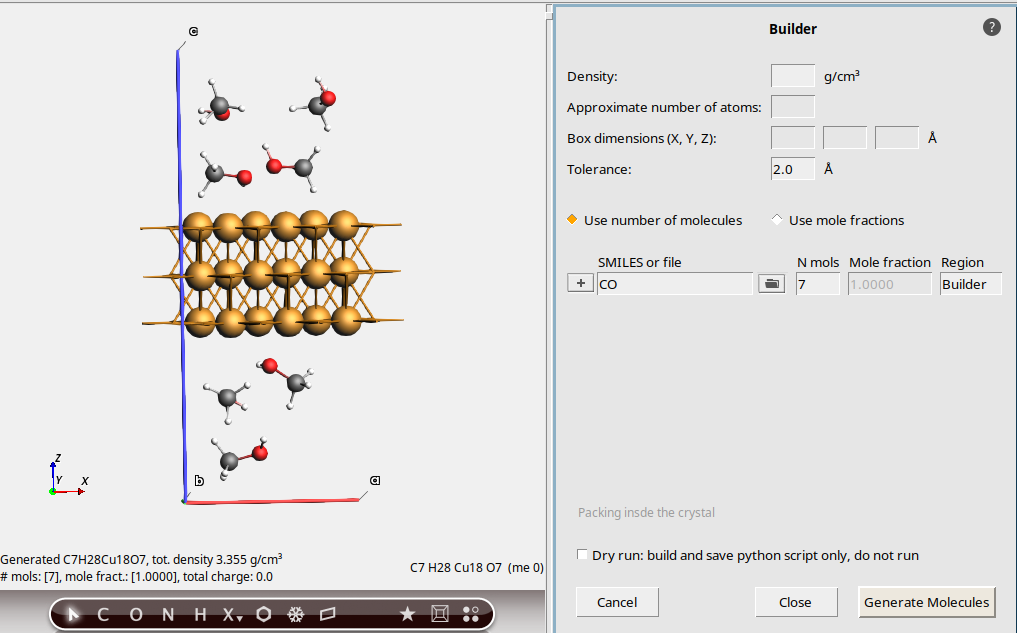
Recommendation: If possible, always specify the number of molecules when packing around a system and do not rely on the estimation of the density.
Builder results are even more approximate for non-orthorhombic unit cells¶
→ System
Atoms
Cu 2.127212899986092 1.473776325712727 12.08423447177468
Cu 2.127212899986094 2.947552654287273 9.999999999999943
Cu 3.403540640027812 2.21066449 7.91576552822538
Cu 3.403540639986093 3.684440815712728 12.08423447177468
Cu 5.956196119986094 0.7368881642872729 9.999999999999943
Cu 7.232523860027812 0.0 7.91576552822538
Cu 4.679868379986093 1.473776325712727 12.08423447177468
Cu 4.679868379986095 2.947552654287273 9.999999999999943
Cu 5.956196120027814 2.21066449 7.91576552822538
Cu -1.701770320013906 3.684440815712728 12.08423447177468
Cu 0.8508851599860943 0.7368881642872729 9.999999999999943
Cu 2.127212900027812 0.0 7.91576552822538
Cu -0.4254425800139062 1.473776325712727 12.08423447177468
Cu -0.4254425800139054 2.947552654287273 9.999999999999943
Cu 0.8508851600278127 2.21066449 7.91576552822538
Cu 0.8508851599860932 3.684440815712728 12.08423447177468
Cu 3.403540639986094 0.7368881642872729 9.999999999999943
Cu 4.679868380027812 0.0 7.91576552822538
End
Lattice
7.657966439999999 0.0 0.0
-2.55265548 4.42132898 0.0
0.0 0.0 20.0
End
End
This is now a hexagonal unit cell, where one of the lattice vector angles is 120 degrees. However, the volume is exactly the same as in the previous case.
CO (SMILES for methanol)Intuitively we would expect to get 7 methanol molecules, just like before.
However, we get fewer than 7.
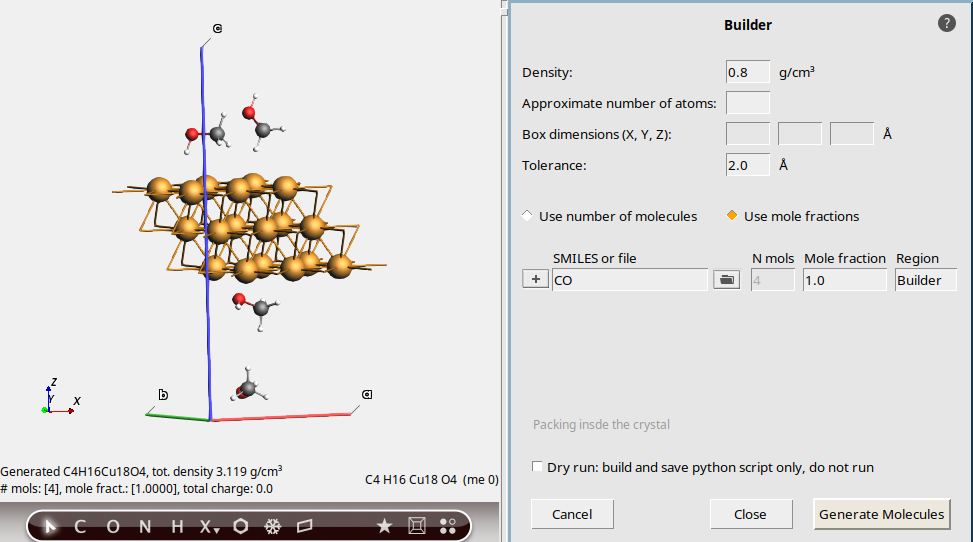
For a non-orthorhombic cell like this, the molecules are packed into a surrounding box, and any molecules that are outside the unit cell are removed. As a result, you typically get fewer molecules packed than for orthorhombic cells.
This is true even if you specify the number of molecules:
7You still get fewer than 7 molecules!
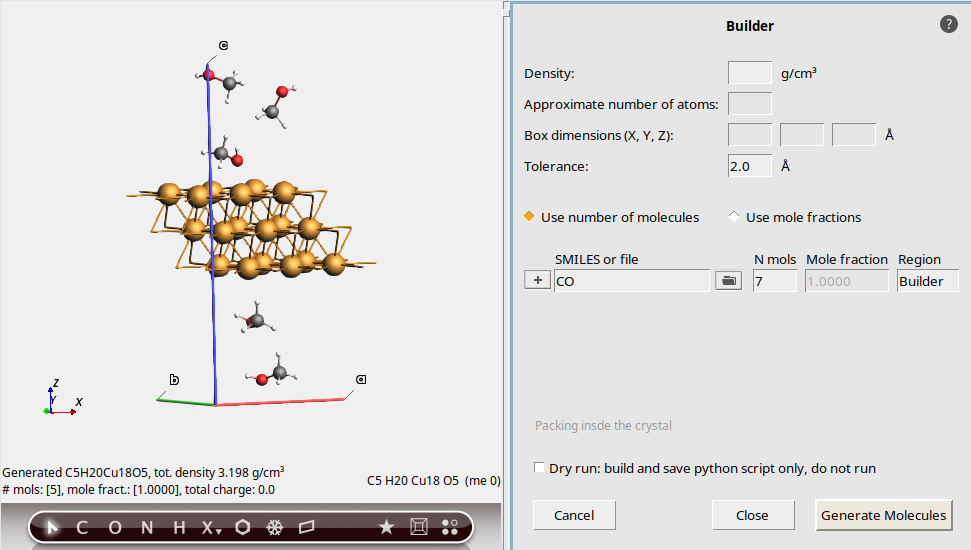
Recommendation: Be extra careful about using the Builder with non-orthorhombic cells. You usually get fewer molecules than you ask for.
Next steps¶
Some features of the Builder are demonstrated in other tutorials.
To pack molecules or ions with specific force field atom types, see Ionic Conductivity.
To set regions for the packed molecules, see Bond boost: Cross-linked polymers.
For a more realistic solid-liquid interface, see Solid-liquid interface
To run the Builder from Python, see the PLAMS packmol function.
More details can be found in the Builder documentation