Ionic Conductivity¶
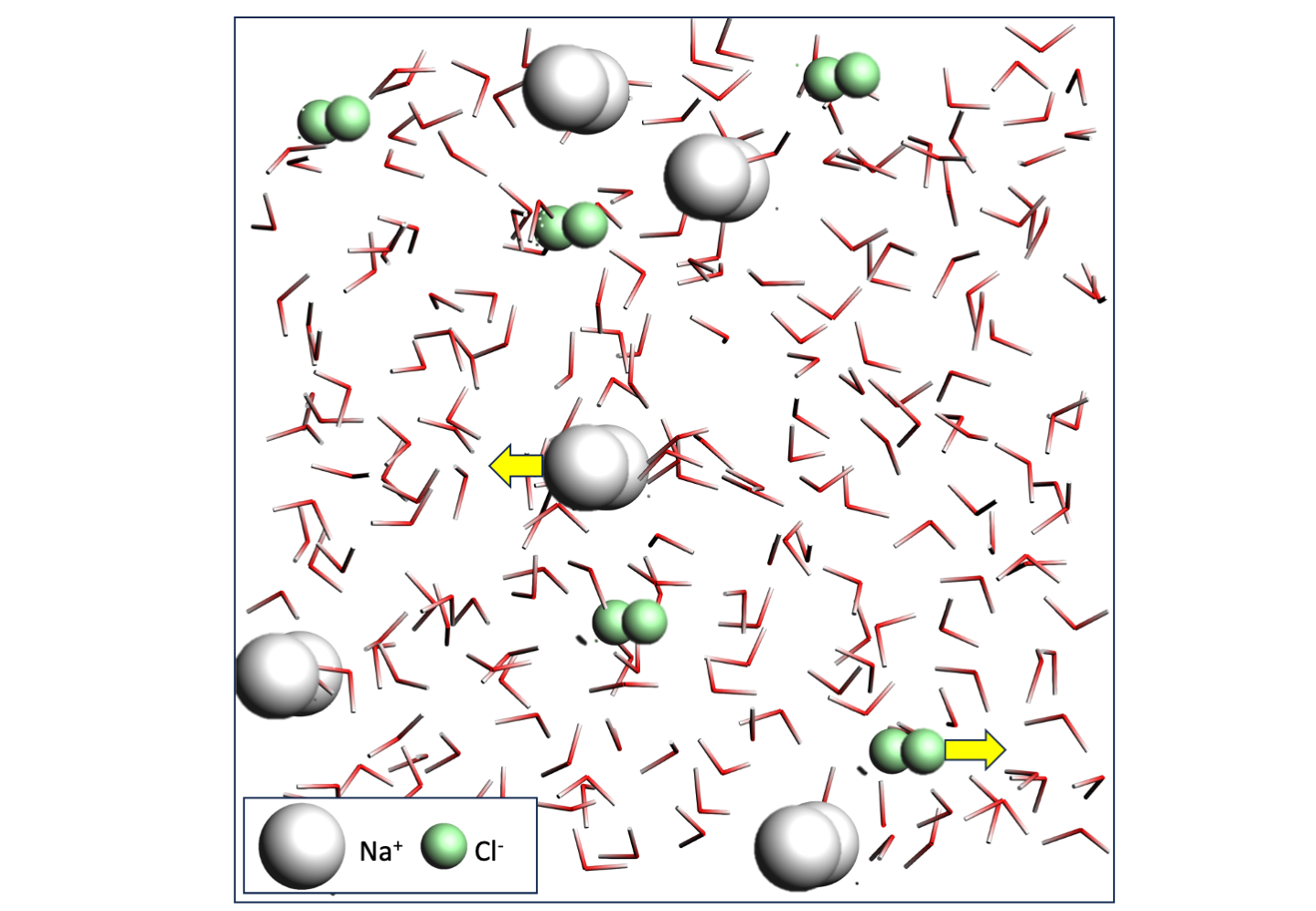
In this tutorial we will compute the ionic conductivity of sodium chloride ions in an aqueous solution by means of molecular dynamics simulations with the ForceField engine.
The system and workflow presented here is very similar to the one described in the publication Estimates of Electrical Conductivity from Molecular Dynamics Simulations: How to Invest the Computational Effort J. Phys. Chem. B 124, 9680−9689 (2020).
Ionic conductivity (S/m) of 1.21 mol/L NaCl in water at 300K |
|
|---|---|
AMS |
7.3 |
Ref. (MD) |
5.9 \(\pm\) 2.9 |
The tutorial consists of the following steps:
Building a sodium chloride solution with AMBER95 atom types.
Equilibrating the system at 300 K
Running a simulation in the NVT ensemble
Obtaining a value for the ionic conductivity
Comparing that value to literature
If you wish, you can download the already-relaxed sodium chloride solution NaCl_wat.in and jump directly to the diffusion coefficient calculation
Building the NaCl solution¶
 →
→ 
First we create the building blocks for our system from scratch.
 next to Atom types
next to Atom types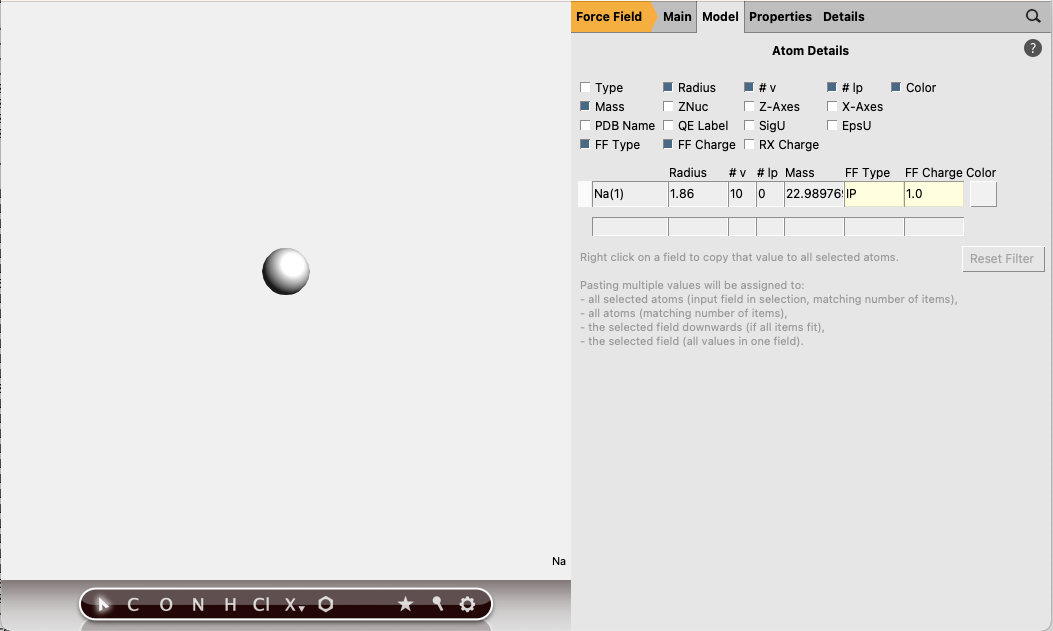
Repeat for a Cl atom (FF Type IM, FF Charge -1.0), and save as “Cl.in”.
Repeat for a single water molecule (FF Types O: OW, both H atoms: HW, FF Charges O: -0.8340, H: 0.4170), and save as “H2O.in”.
Na.in
System
Atoms
Na 0.0 0.0 0.0 ForceField.Type=IP ForceField.Charge=1.0
End
End
Cl.in
System
Atoms
Cl 0.0 0.0 0.0 ForceField.Type=IM ForceField.Charge=-1.0
End
End
H2O.in
System
Atoms
O 0.0 0.0 0.40468149 ForceField.Type=OW ForceField.Charge=-0.834
H 0.78258644 0.0 -0.20234075 ForceField.Type=HW ForceField.Charge=0.417
H -0.78258644 0.0 -0.20234075 ForceField.Type=HW ForceField.Charge=0.417
End
BondOrders
1 2 1.0
1 3 1.0
End
End
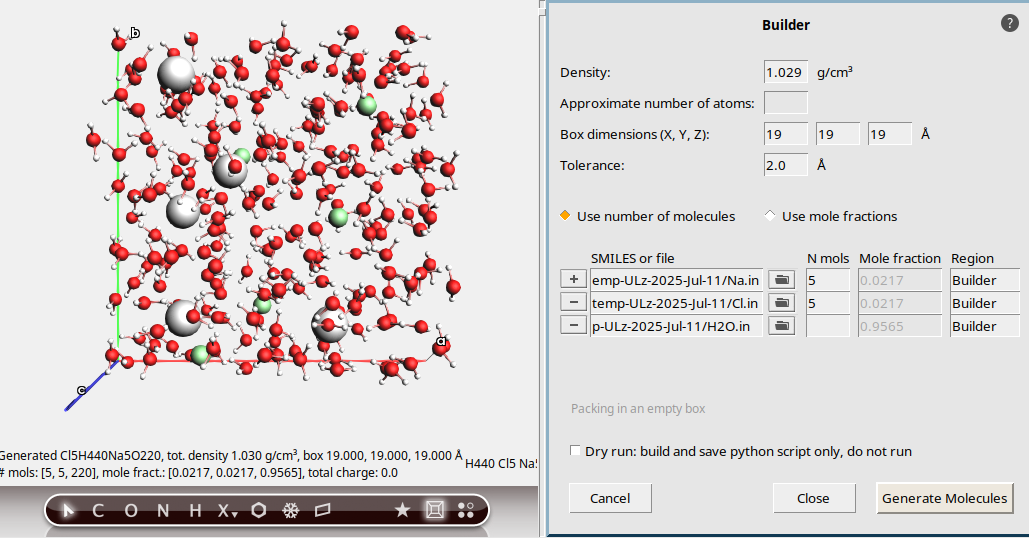
Now we are ready to build the full system.
Important
While building the system, make sure that the Amber95 force field is still selected. Changing the ForceField Type after building will result in all atom typing information being erased.
19 19 19 Å.5 to add a new molecule
to add a new molecule5 to add a new moleculeThis should result in an aqueous NaCl solution with a density of 1.029 g/mL, containing 5 Na⁺, 5 Cl⁻, and 220 water molecules.
Note
New in AMS2026: The possibility to automatically determine the number of water molecules was introduced in AMS2026. For previous AMS versions, set the number of water molecules to 220 and do not set the density.
Equilibrating the system¶
First perform a geometry optimization.

Equilibrate the resulting system at the desired temperature
next to Task: Molecular Dynamics to go to the MD details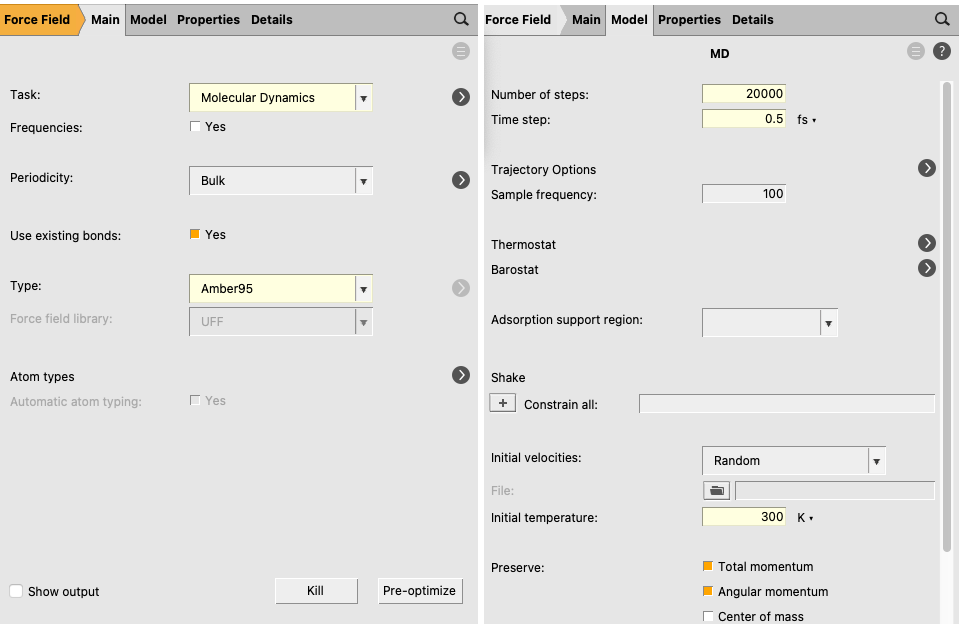 next to Thermostat to go to the thermostat details
next to Thermostat to go to the thermostat details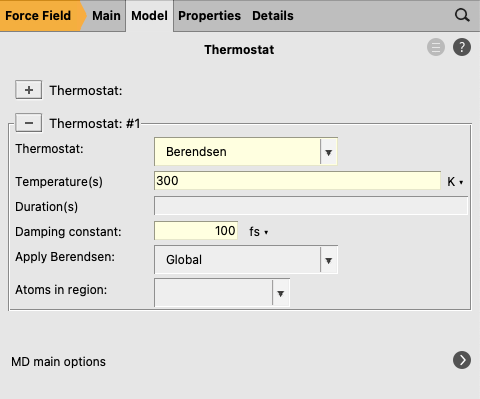
You are now ready to run the simulation.
AMSMovie can plot the potential energy of the system over the course of the simulation.
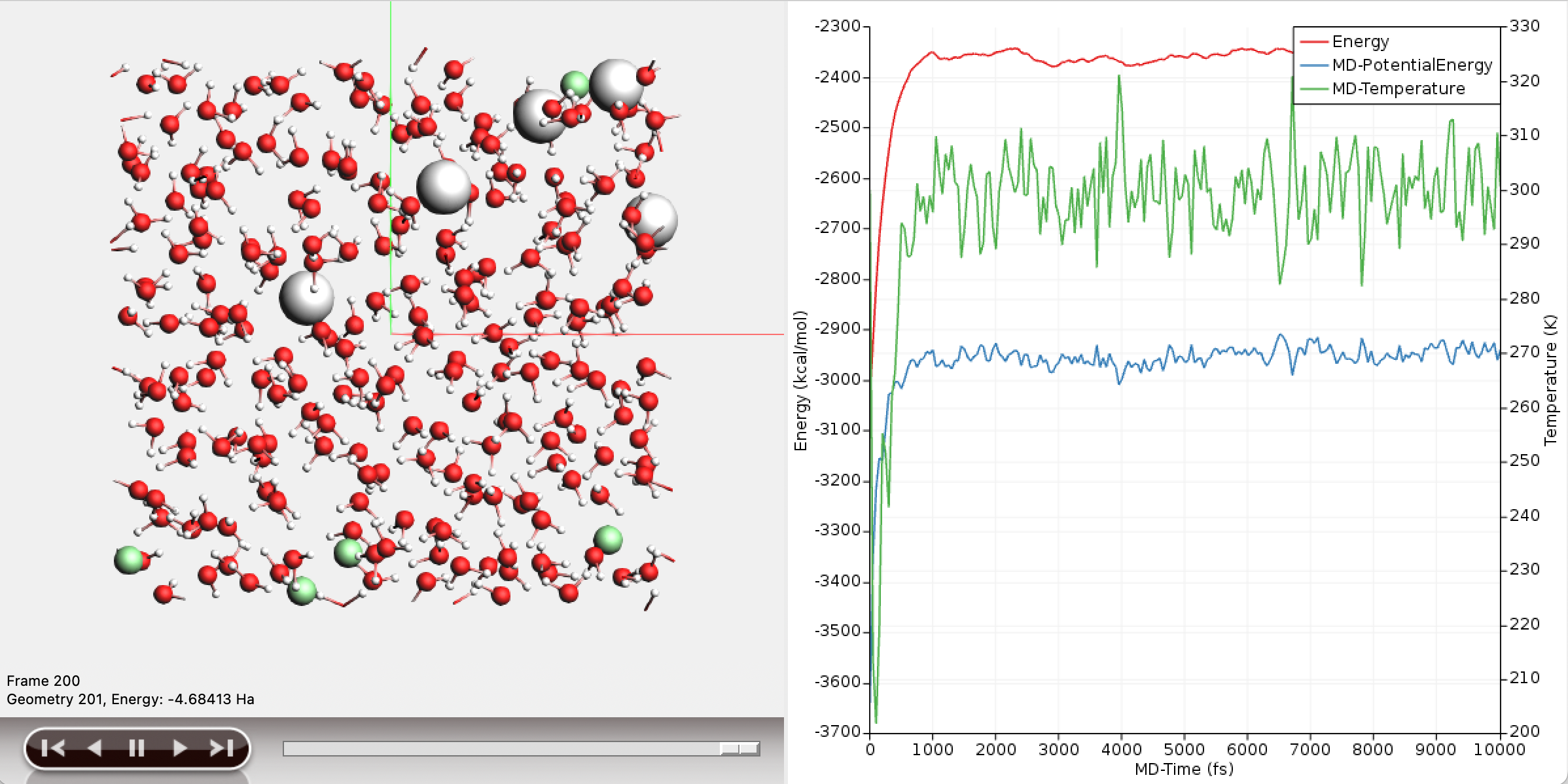
The potential energy of the system has converged to a stable value over the course of the simulation, and the temperature oscillates around 300K.
Running the Production Simulation¶
When the equilibration calculation has finished:
We will run the production simulation using a Nose-Hoover chain thermostat.
next to Task: Molecular Dynamics to go to the MD detailsIf you ran the equilibration yourself:
Otherwise, if you simply imported the provided equilibrated structure:
300Then continue with the thermostat settings:
next to Thermostat to go to the thermostat detailsYou are now ready to run the simulation.
Wait for the calculation to finish (this may take around 45 minutes, depending on your hardware).
Computing the Ionic Conductivity¶
The ionic conductivity is proportional to the slope of the charge weighted mean square displacement of the atoms.
\(\sigma = \lim_{t\to\infty} \frac{1}{2tdVk_BT} \sum_i^N q_i^2 \langle ({\bf{r_i}}(0) - {\bf{r_i}}(t))^2 \rangle\)
Here \(d\) is the dimensionality of the systems (usually \(d=3\)), and \(N\) is the number of ions.
To compute it, open the completed simulation in AMSMovie.
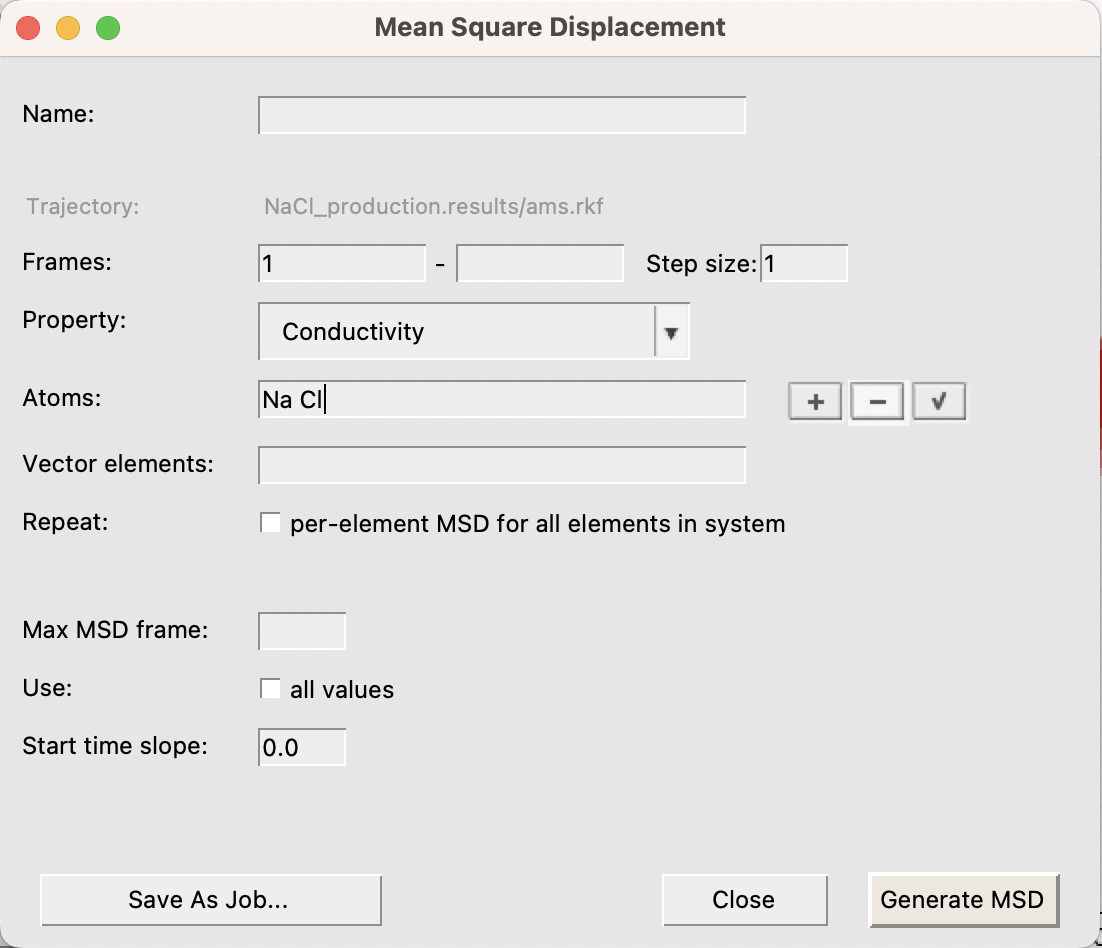
Important
The example simulation presented here is too short to obtain really reliable results. In practice it is advised to perform multiple simulations of several nanoseconds each. Your results may differ by a factor of 3 to the results presented here.
Two new curves have appeared in the AMSMovie window. The bottom curve depicts the convergence of the ionic conductivity with the correlation time. The final value of the ionic conductivity is 7.34 S/m (or C²/Jms). The corresponding literature states that the value should fall between 1.2 and 13.5 S/m, with an average over many simulations of 5.9 S/m.
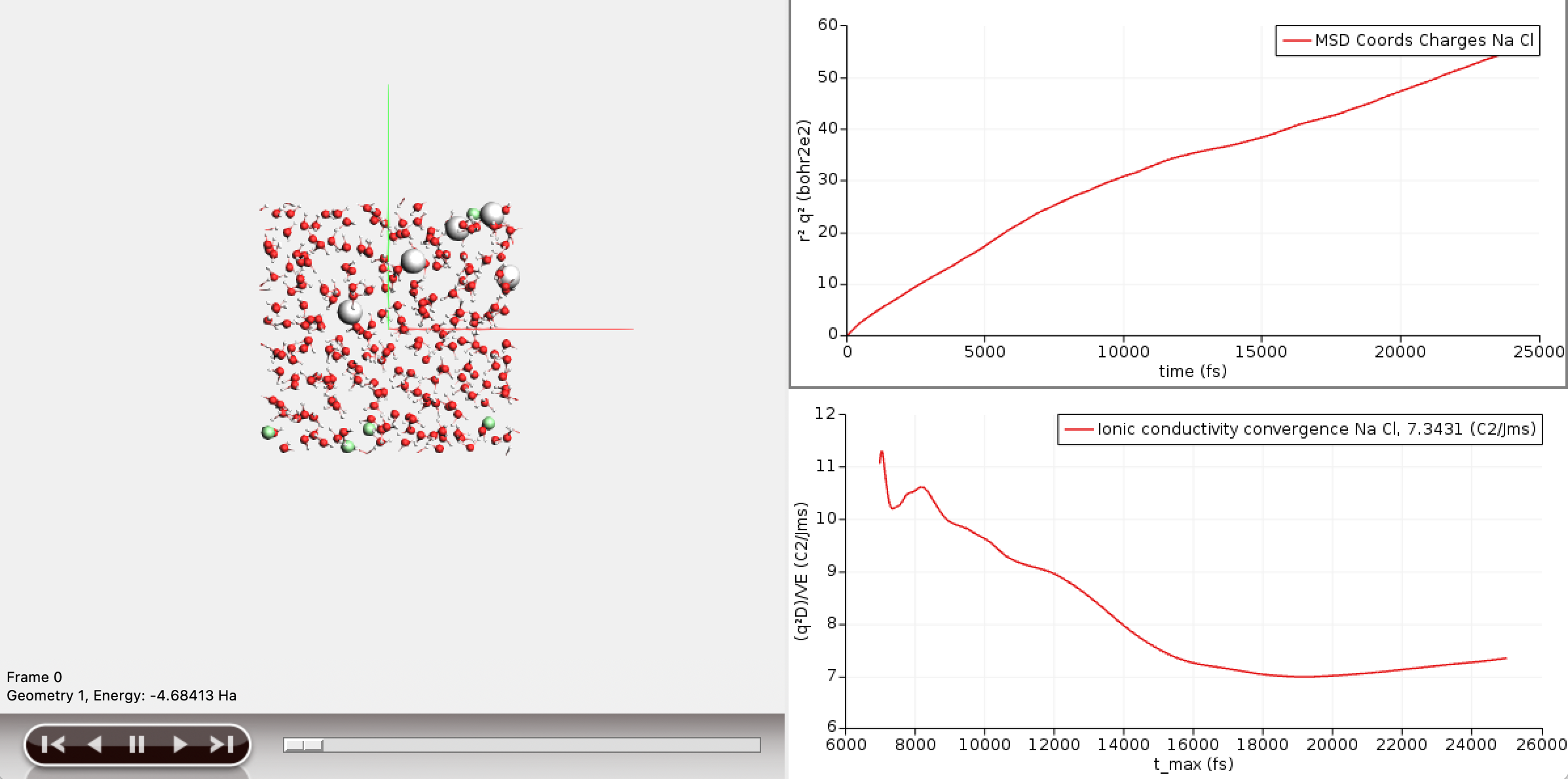
The top curve depicts the charge weighted mean square displacement curve, of which the ionic conductivity is the slope. The graph shows that at short correlation times the slope of the curve is large, and then gradually becomes constant. The point where the curve becomes linear roughly corresponds to the time it takes for the motions of the ions to decorrelate. AMSMovie attempts to estimate this point from the shape of the mean square displacement curve. Indeed, the bottom graph starts at around 7ps, which is AMSMovies guess. However, a brief look at the top curve suggests that a better guess for the decorrelation time would be around 10 ps. We can recompute the ionic conductivity with this new decorrelation time in mind.
The new value for the ionic conductivity is 7.29 S/m.
Important
The publication J. Phys. Chem. B 124, 9680−9689 (2020) discusses how the exact, correlated, ionic conductivity would include off-diagonal contributions to the mean square displacement.
\(\sigma_{corr} = \lim_{t\to\infty} \frac{1}{2tdVk_BT} \left( \sum_i^N q_i^2 \langle ({\bf{r_i}}(0) - {\bf{r_i}}(t))^2 \rangle + \sum_{i \neq j}^N q_i q_j \langle ({\bf{r_i}}(0) - {\bf{r_i}}(t)) ({\bf{r_j}}(0) - {\bf{r_j}}(t)) \rangle \right)\)
In this example we used high NaCl concentrations to be able to compare with literature values. Indeed, at these concentrations, the off-diagonal contributions can change the value of \(\sigma\) by approximately a factor two. At lower ion concentrations the effect of the off-diagonal contributions will become negligible.
Important
AMSMovie uses the atomic charges as they are stored on the ams.rkf file (in this case the forcefield charges). In this example, the forcefield charges match the formal charges, but for larger ions, this may not be the case, and this will affect the results.