Solid-liquid interface¶
This tutorial will help you to:
create an aluminum slab
add a solvent (water)
set different thermostats (temperatures) for different regions of the system.
run the molecular dynamics simulation and see what happens
For this simulation we will use the ReaxFF force field through the AMS driver program.
Note
The purpose of this tutorial is to demonstrate how to technically set up and run a simulation. The employed Al-H2O ReaxFF force field was parameterized to handle similar, but not exactly the same, types of systems as in this tutorial. Always do your own testing to see if the results from a simulation are reasonable.
Step 1: Start AMSinput in ReaxFF mode¶
 →
→ 
Step 2: Creating the surface¶
To create the surface, we will
Get the primitive bulk Al unit cell
Convert the primitive cell to conventional cell
Create the surface
Create a 9x9 supercell
Define a z dimension of 50 Å
Center the slab in the cell
Tip
If you’re already familiar with building crystals and slabs, copy-paste the contents of the below box into AMSinput to skip directly to Step 3.
Copy-paste system to skip to Step 3
System
Atoms
Al 32.2176 32.2176 28.0375
Al 33.6494 33.6494 26.0125
Al 32.2176 32.2176 23.9875
Al 33.6494 33.6494 21.9625
Al 0.7159 0.7159 28.0375
Al 2.1478 2.1478 26.0125
Al 0.7159 0.7159 23.9875
Al 2.1478 2.1478 21.9625
Al 0.7159 3.5797 28.0375
Al 2.1478 5.0116 26.0125
Al 0.7159 3.5797 23.9875
Al 2.1478 5.0116 21.9625
Al 0.7159 6.4435 28.0375
Al 2.1478 7.8754 26.0125
Al 0.7159 6.4435 23.9875
Al 2.1478 7.8754 21.9625
Al 0.7159 9.3073 28.0375
Al 2.1478 10.7392 26.0125
Al 0.7159 9.3073 23.9875
Al 2.1478 10.7392 21.9625
Al 0.7159 12.1711 28.0375
Al 2.1478 13.6030 26.0125
Al 0.7159 12.1711 23.9875
Al 2.1478 13.6030 21.9625
Al 0.7159 15.0349 28.0375
Al 2.1478 16.4667 26.0125
Al 0.7159 15.0349 23.9875
Al 2.1478 16.4667 21.9625
Al 0.7159 17.8986 28.0375
Al 2.1478 19.3305 26.0125
Al 0.7159 17.8986 23.9875
Al 2.1478 19.3305 21.9625
Al 0.7159 20.7624 28.0375
Al 2.1478 22.1943 26.0125
Al 0.7159 20.7624 23.9875
Al 2.1478 22.1943 21.9625
Al 0.7159 23.6262 28.0375
Al 2.1478 25.0581 26.0125
Al 0.7159 23.6262 23.9875
Al 2.1478 25.0581 21.9625
Al 0.7159 26.4900 28.0375
Al 2.1478 27.9219 26.0125
Al 0.7159 26.4900 23.9875
Al 2.1478 27.9219 21.9625
Al 0.7159 29.3538 28.0375
Al 2.1478 30.7857 26.0125
Al 0.7159 29.3538 23.9875
Al 2.1478 30.7857 21.9625
Al 0.7159 32.2176 28.0375
Al 2.1478 33.6494 26.0125
Al 0.7159 32.2176 23.9875
Al 2.1478 33.6494 21.9625
Al 3.5797 0.7159 28.0375
Al 5.0116 2.1478 26.0125
Al 3.5797 0.7159 23.9875
Al 5.0116 2.1478 21.9625
Al 3.5797 3.5797 28.0375
Al 5.0116 5.0116 26.0125
Al 3.5797 3.5797 23.9875
Al 5.0116 5.0116 21.9625
Al 3.5797 6.4435 28.0375
Al 5.0116 7.8754 26.0125
Al 3.5797 6.4435 23.9875
Al 5.0116 7.8754 21.9625
Al 3.5797 9.3073 28.0375
Al 5.0116 10.7392 26.0125
Al 3.5797 9.3073 23.9875
Al 5.0116 10.7392 21.9625
Al 3.5797 12.1711 28.0375
Al 5.0116 13.6030 26.0125
Al 3.5797 12.1711 23.9875
Al 5.0116 13.6030 21.9625
Al 3.5797 15.0349 28.0375
Al 5.0116 16.4667 26.0125
Al 3.5797 15.0349 23.9875
Al 5.0116 16.4667 21.9625
Al 3.5797 17.8986 28.0375
Al 5.0116 19.3305 26.0125
Al 3.5797 17.8986 23.9875
Al 5.0116 19.3305 21.9625
Al 3.5797 20.7624 28.0375
Al 5.0116 22.1943 26.0125
Al 3.5797 20.7624 23.9875
Al 5.0116 22.1943 21.9625
Al 3.5797 23.6262 28.0375
Al 5.0116 25.0581 26.0125
Al 3.5797 23.6262 23.9875
Al 5.0116 25.0581 21.9625
Al 3.5797 26.4900 28.0375
Al 5.0116 27.9219 26.0125
Al 3.5797 26.4900 23.9875
Al 5.0116 27.9219 21.9625
Al 3.5797 29.3538 28.0375
Al 5.0116 30.7857 26.0125
Al 3.5797 29.3538 23.9875
Al 5.0116 30.7857 21.9625
Al 3.5797 32.2176 28.0375
Al 5.0116 33.6494 26.0125
Al 3.5797 32.2176 23.9875
Al 5.0116 33.6494 21.9625
Al 6.4435 0.7159 28.0375
Al 7.8754 2.1478 26.0125
Al 6.4435 0.7159 23.9875
Al 7.8754 2.1478 21.9625
Al 6.4435 3.5797 28.0375
Al 7.8754 5.0116 26.0125
Al 6.4435 3.5797 23.9875
Al 7.8754 5.0116 21.9625
Al 6.4435 6.4435 28.0375
Al 7.8754 7.8754 26.0125
Al 6.4435 6.4435 23.9875
Al 7.8754 7.8754 21.9625
Al 6.4435 9.3073 28.0375
Al 7.8754 10.7392 26.0125
Al 6.4435 9.3073 23.9875
Al 7.8754 10.7392 21.9625
Al 6.4435 12.1711 28.0375
Al 7.8754 13.6030 26.0125
Al 6.4435 12.1711 23.9875
Al 7.8754 13.6030 21.9625
Al 6.4435 15.0349 28.0375
Al 7.8754 16.4667 26.0125
Al 6.4435 15.0349 23.9875
Al 7.8754 16.4667 21.9625
Al 6.4435 17.8986 28.0375
Al 7.8754 19.3305 26.0125
Al 6.4435 17.8986 23.9875
Al 7.8754 19.3305 21.9625
Al 6.4435 20.7624 28.0375
Al 7.8754 22.1943 26.0125
Al 6.4435 20.7624 23.9875
Al 7.8754 22.1943 21.9625
Al 6.4435 23.6262 28.0375
Al 7.8754 25.0581 26.0125
Al 6.4435 23.6262 23.9875
Al 7.8754 25.0581 21.9625
Al 6.4435 26.4900 28.0375
Al 7.8754 27.9219 26.0125
Al 6.4435 26.4900 23.9875
Al 7.8754 27.9219 21.9625
Al 6.4435 29.3538 28.0375
Al 7.8754 30.7857 26.0125
Al 6.4435 29.3538 23.9875
Al 7.8754 30.7857 21.9625
Al 6.4435 32.2176 28.0375
Al 7.8754 33.6494 26.0125
Al 6.4435 32.2176 23.9875
Al 7.8754 33.6494 21.9625
Al 9.3073 0.7159 28.0375
Al 10.7392 2.1478 26.0125
Al 9.3073 0.7159 23.9875
Al 10.7392 2.1478 21.9625
Al 9.3073 3.5797 28.0375
Al 10.7392 5.0116 26.0125
Al 9.3073 3.5797 23.9875
Al 10.7392 5.0116 21.9625
Al 9.3073 6.4435 28.0375
Al 10.7392 7.8754 26.0125
Al 9.3073 6.4435 23.9875
Al 10.7392 7.8754 21.9625
Al 9.3073 9.3073 28.0375
Al 10.7392 10.7392 26.0125
Al 9.3073 9.3073 23.9875
Al 10.7392 10.7392 21.9625
Al 9.3073 12.1711 28.0375
Al 10.7392 13.6030 26.0125
Al 9.3073 12.1711 23.9875
Al 10.7392 13.6030 21.9625
Al 9.3073 15.0349 28.0375
Al 10.7392 16.4667 26.0125
Al 9.3073 15.0349 23.9875
Al 10.7392 16.4667 21.9625
Al 9.3073 17.8986 28.0375
Al 10.7392 19.3305 26.0125
Al 9.3073 17.8986 23.9875
Al 10.7392 19.3305 21.9625
Al 9.3073 20.7624 28.0375
Al 10.7392 22.1943 26.0125
Al 9.3073 20.7624 23.9875
Al 10.7392 22.1943 21.9625
Al 9.3073 23.6262 28.0375
Al 10.7392 25.0581 26.0125
Al 9.3073 23.6262 23.9875
Al 10.7392 25.0581 21.9625
Al 9.3073 26.4900 28.0375
Al 10.7392 27.9219 26.0125
Al 9.3073 26.4900 23.9875
Al 10.7392 27.9219 21.9625
Al 9.3073 29.3538 28.0375
Al 10.7392 30.7857 26.0125
Al 9.3073 29.3538 23.9875
Al 10.7392 30.7857 21.9625
Al 9.3073 32.2176 28.0375
Al 10.7392 33.6494 26.0125
Al 9.3073 32.2176 23.9875
Al 10.7392 33.6494 21.9625
Al 12.1711 0.7159 28.0375
Al 13.6030 2.1478 26.0125
Al 12.1711 0.7159 23.9875
Al 13.6030 2.1478 21.9625
Al 12.1711 3.5797 28.0375
Al 13.6030 5.0116 26.0125
Al 12.1711 3.5797 23.9875
Al 13.6030 5.0116 21.9625
Al 12.1711 6.4435 28.0375
Al 13.6030 7.8754 26.0125
Al 12.1711 6.4435 23.9875
Al 13.6030 7.8754 21.9625
Al 12.1711 9.3073 28.0375
Al 13.6030 10.7392 26.0125
Al 12.1711 9.3073 23.9875
Al 13.6030 10.7392 21.9625
Al 12.1711 12.1711 28.0375
Al 13.6030 13.6030 26.0125
Al 12.1711 12.1711 23.9875
Al 13.6030 13.6030 21.9625
Al 12.1711 15.0349 28.0375
Al 13.6030 16.4667 26.0125
Al 12.1711 15.0349 23.9875
Al 13.6030 16.4667 21.9625
Al 12.1711 17.8986 28.0375
Al 13.6030 19.3305 26.0125
Al 12.1711 17.8986 23.9875
Al 13.6030 19.3305 21.9625
Al 12.1711 20.7624 28.0375
Al 13.6030 22.1943 26.0125
Al 12.1711 20.7624 23.9875
Al 13.6030 22.1943 21.9625
Al 12.1711 23.6262 28.0375
Al 13.6030 25.0581 26.0125
Al 12.1711 23.6262 23.9875
Al 13.6030 25.0581 21.9625
Al 12.1711 26.4900 28.0375
Al 13.6030 27.9219 26.0125
Al 12.1711 26.4900 23.9875
Al 13.6030 27.9219 21.9625
Al 12.1711 29.3538 28.0375
Al 13.6030 30.7857 26.0125
Al 12.1711 29.3538 23.9875
Al 13.6030 30.7857 21.9625
Al 12.1711 32.2176 28.0375
Al 13.6030 33.6494 26.0125
Al 12.1711 32.2176 23.9875
Al 13.6030 33.6494 21.9625
Al 15.0349 0.7159 28.0375
Al 16.4667 2.1478 26.0125
Al 15.0349 0.7159 23.9875
Al 16.4667 2.1478 21.9625
Al 15.0349 3.5797 28.0375
Al 16.4667 5.0116 26.0125
Al 15.0349 3.5797 23.9875
Al 16.4667 5.0116 21.9625
Al 15.0349 6.4435 28.0375
Al 16.4667 7.8754 26.0125
Al 15.0349 6.4435 23.9875
Al 16.4667 7.8754 21.9625
Al 15.0349 9.3073 28.0375
Al 16.4667 10.7392 26.0125
Al 15.0349 9.3073 23.9875
Al 16.4667 10.7392 21.9625
Al 15.0349 12.1711 28.0375
Al 16.4667 13.6030 26.0125
Al 15.0349 12.1711 23.9875
Al 16.4667 13.6030 21.9625
Al 15.0349 15.0349 28.0375
Al 16.4667 16.4667 26.0125
Al 15.0349 15.0349 23.9875
Al 16.4667 16.4667 21.9625
Al 15.0349 17.8986 28.0375
Al 16.4667 19.3305 26.0125
Al 15.0349 17.8986 23.9875
Al 16.4667 19.3305 21.9625
Al 15.0349 20.7624 28.0375
Al 16.4667 22.1943 26.0125
Al 15.0349 20.7624 23.9875
Al 16.4667 22.1943 21.9625
Al 15.0349 23.6262 28.0375
Al 16.4667 25.0581 26.0125
Al 15.0349 23.6262 23.9875
Al 16.4667 25.0581 21.9625
Al 15.0349 26.4900 28.0375
Al 16.4667 27.9219 26.0125
Al 15.0349 26.4900 23.9875
Al 16.4667 27.9219 21.9625
Al 15.0349 29.3538 28.0375
Al 16.4667 30.7857 26.0125
Al 15.0349 29.3538 23.9875
Al 16.4667 30.7857 21.9625
Al 15.0349 32.2176 28.0375
Al 16.4667 33.6494 26.0125
Al 15.0349 32.2176 23.9875
Al 16.4667 33.6494 21.9625
Al 17.8986 0.7159 28.0375
Al 19.3305 2.1478 26.0125
Al 17.8986 0.7159 23.9875
Al 19.3305 2.1478 21.9625
Al 17.8986 3.5797 28.0375
Al 19.3305 5.0116 26.0125
Al 17.8986 3.5797 23.9875
Al 19.3305 5.0116 21.9625
Al 17.8986 6.4435 28.0375
Al 19.3305 7.8754 26.0125
Al 17.8986 6.4435 23.9875
Al 19.3305 7.8754 21.9625
Al 17.8986 9.3073 28.0375
Al 19.3305 10.7392 26.0125
Al 17.8986 9.3073 23.9875
Al 19.3305 10.7392 21.9625
Al 17.8986 12.1711 28.0375
Al 19.3305 13.6030 26.0125
Al 17.8986 12.1711 23.9875
Al 19.3305 13.6030 21.9625
Al 17.8986 15.0349 28.0375
Al 19.3305 16.4667 26.0125
Al 17.8986 15.0349 23.9875
Al 19.3305 16.4667 21.9625
Al 17.8986 17.8986 28.0375
Al 19.3305 19.3305 26.0125
Al 17.8986 17.8986 23.9875
Al 19.3305 19.3305 21.9625
Al 17.8986 20.7624 28.0375
Al 19.3305 22.1943 26.0125
Al 17.8986 20.7624 23.9875
Al 19.3305 22.1943 21.9625
Al 17.8986 23.6262 28.0375
Al 19.3305 25.0581 26.0125
Al 17.8986 23.6262 23.9875
Al 19.3305 25.0581 21.9625
Al 17.8986 26.4900 28.0375
Al 19.3305 27.9219 26.0125
Al 17.8986 26.4900 23.9875
Al 19.3305 27.9219 21.9625
Al 17.8986 29.3538 28.0375
Al 19.3305 30.7857 26.0125
Al 17.8986 29.3538 23.9875
Al 19.3305 30.7857 21.9625
Al 17.8986 32.2176 28.0375
Al 19.3305 33.6494 26.0125
Al 17.8986 32.2176 23.9875
Al 19.3305 33.6494 21.9625
Al 20.7624 0.7159 28.0375
Al 22.1943 2.1478 26.0125
Al 20.7624 0.7159 23.9875
Al 22.1943 2.1478 21.9625
Al 20.7624 3.5797 28.0375
Al 22.1943 5.0116 26.0125
Al 20.7624 3.5797 23.9875
Al 22.1943 5.0116 21.9625
Al 20.7624 6.4435 28.0375
Al 22.1943 7.8754 26.0125
Al 20.7624 6.4435 23.9875
Al 22.1943 7.8754 21.9625
Al 20.7624 9.3073 28.0375
Al 22.1943 10.7392 26.0125
Al 20.7624 9.3073 23.9875
Al 22.1943 10.7392 21.9625
Al 20.7624 12.1711 28.0375
Al 22.1943 13.6030 26.0125
Al 20.7624 12.1711 23.9875
Al 22.1943 13.6030 21.9625
Al 20.7624 15.0349 28.0375
Al 22.1943 16.4667 26.0125
Al 20.7624 15.0349 23.9875
Al 22.1943 16.4667 21.9625
Al 20.7624 17.8986 28.0375
Al 22.1943 19.3305 26.0125
Al 20.7624 17.8986 23.9875
Al 22.1943 19.3305 21.9625
Al 20.7624 20.7624 28.0375
Al 22.1943 22.1943 26.0125
Al 20.7624 20.7624 23.9875
Al 22.1943 22.1943 21.9625
Al 20.7624 23.6262 28.0375
Al 22.1943 25.0581 26.0125
Al 20.7624 23.6262 23.9875
Al 22.1943 25.0581 21.9625
Al 20.7624 26.4900 28.0375
Al 22.1943 27.9219 26.0125
Al 20.7624 26.4900 23.9875
Al 22.1943 27.9219 21.9625
Al 20.7624 29.3538 28.0375
Al 22.1943 30.7857 26.0125
Al 20.7624 29.3538 23.9875
Al 22.1943 30.7857 21.9625
Al 20.7624 32.2176 28.0375
Al 22.1943 33.6494 26.0125
Al 20.7624 32.2176 23.9875
Al 22.1943 33.6494 21.9625
Al 23.6262 0.7159 28.0375
Al 25.0581 2.1478 26.0125
Al 23.6262 0.7159 23.9875
Al 25.0581 2.1478 21.9625
Al 23.6262 3.5797 28.0375
Al 25.0581 5.0116 26.0125
Al 23.6262 3.5797 23.9875
Al 25.0581 5.0116 21.9625
Al 23.6262 6.4435 28.0375
Al 25.0581 7.8754 26.0125
Al 23.6262 6.4435 23.9875
Al 25.0581 7.8754 21.9625
Al 23.6262 9.3073 28.0375
Al 25.0581 10.7392 26.0125
Al 23.6262 9.3073 23.9875
Al 25.0581 10.7392 21.9625
Al 23.6262 12.1711 28.0375
Al 25.0581 13.6030 26.0125
Al 23.6262 12.1711 23.9875
Al 25.0581 13.6030 21.9625
Al 23.6262 15.0349 28.0375
Al 25.0581 16.4667 26.0125
Al 23.6262 15.0349 23.9875
Al 25.0581 16.4667 21.9625
Al 23.6262 17.8986 28.0375
Al 25.0581 19.3305 26.0125
Al 23.6262 17.8986 23.9875
Al 25.0581 19.3305 21.9625
Al 23.6262 20.7624 28.0375
Al 25.0581 22.1943 26.0125
Al 23.6262 20.7624 23.9875
Al 25.0581 22.1943 21.9625
Al 23.6262 23.6262 28.0375
Al 25.0581 25.0581 26.0125
Al 23.6262 23.6262 23.9875
Al 25.0581 25.0581 21.9625
Al 23.6262 26.4900 28.0375
Al 25.0581 27.9219 26.0125
Al 23.6262 26.4900 23.9875
Al 25.0581 27.9219 21.9625
Al 23.6262 29.3538 28.0375
Al 25.0581 30.7857 26.0125
Al 23.6262 29.3538 23.9875
Al 25.0581 30.7857 21.9625
Al 23.6262 32.2176 28.0375
Al 25.0581 33.6494 26.0125
Al 23.6262 32.2176 23.9875
Al 25.0581 33.6494 21.9625
Al 26.4900 0.7159 28.0375
Al 27.9219 2.1478 26.0125
Al 26.4900 0.7159 23.9875
Al 27.9219 2.1478 21.9625
Al 26.4900 3.5797 28.0375
Al 27.9219 5.0116 26.0125
Al 26.4900 3.5797 23.9875
Al 27.9219 5.0116 21.9625
Al 26.4900 6.4435 28.0375
Al 27.9219 7.8754 26.0125
Al 26.4900 6.4435 23.9875
Al 27.9219 7.8754 21.9625
Al 26.4900 9.3073 28.0375
Al 27.9219 10.7392 26.0125
Al 26.4900 9.3073 23.9875
Al 27.9219 10.7392 21.9625
Al 26.4900 12.1711 28.0375
Al 27.9219 13.6030 26.0125
Al 26.4900 12.1711 23.9875
Al 27.9219 13.6030 21.9625
Al 26.4900 15.0349 28.0375
Al 27.9219 16.4667 26.0125
Al 26.4900 15.0349 23.9875
Al 27.9219 16.4667 21.9625
Al 26.4900 17.8986 28.0375
Al 27.9219 19.3305 26.0125
Al 26.4900 17.8986 23.9875
Al 27.9219 19.3305 21.9625
Al 26.4900 20.7624 28.0375
Al 27.9219 22.1943 26.0125
Al 26.4900 20.7624 23.9875
Al 27.9219 22.1943 21.9625
Al 26.4900 23.6262 28.0375
Al 27.9219 25.0581 26.0125
Al 26.4900 23.6262 23.9875
Al 27.9219 25.0581 21.9625
Al 26.4900 26.4900 28.0375
Al 27.9219 27.9219 26.0125
Al 26.4900 26.4900 23.9875
Al 27.9219 27.9219 21.9625
Al 26.4900 29.3538 28.0375
Al 27.9219 30.7857 26.0125
Al 26.4900 29.3538 23.9875
Al 27.9219 30.7857 21.9625
Al 26.4900 32.2176 28.0375
Al 27.9219 33.6494 26.0125
Al 26.4900 32.2176 23.9875
Al 27.9219 33.6494 21.9625
Al 29.3538 0.7159 28.0375
Al 30.7857 2.1478 26.0125
Al 29.3538 0.7159 23.9875
Al 30.7857 2.1478 21.9625
Al 29.3538 3.5797 28.0375
Al 30.7857 5.0116 26.0125
Al 29.3538 3.5797 23.9875
Al 30.7857 5.0116 21.9625
Al 29.3538 6.4435 28.0375
Al 30.7857 7.8754 26.0125
Al 29.3538 6.4435 23.9875
Al 30.7857 7.8754 21.9625
Al 29.3538 9.3073 28.0375
Al 30.7857 10.7392 26.0125
Al 29.3538 9.3073 23.9875
Al 30.7857 10.7392 21.9625
Al 29.3538 12.1711 28.0375
Al 30.7857 13.6030 26.0125
Al 29.3538 12.1711 23.9875
Al 30.7857 13.6030 21.9625
Al 29.3538 15.0349 28.0375
Al 30.7857 16.4667 26.0125
Al 29.3538 15.0349 23.9875
Al 30.7857 16.4667 21.9625
Al 29.3538 17.8986 28.0375
Al 30.7857 19.3305 26.0125
Al 29.3538 17.8986 23.9875
Al 30.7857 19.3305 21.9625
Al 29.3538 20.7624 28.0375
Al 30.7857 22.1943 26.0125
Al 29.3538 20.7624 23.9875
Al 30.7857 22.1943 21.9625
Al 29.3538 23.6262 28.0375
Al 30.7857 25.0581 26.0125
Al 29.3538 23.6262 23.9875
Al 30.7857 25.0581 21.9625
Al 29.3538 26.4900 28.0375
Al 30.7857 27.9219 26.0125
Al 29.3538 26.4900 23.9875
Al 30.7857 27.9219 21.9625
Al 29.3538 29.3538 28.0375
Al 30.7857 30.7857 26.0125
Al 29.3538 29.3538 23.9875
Al 30.7857 30.7857 21.9625
Al 29.3538 32.2176 28.0375
Al 30.7857 33.6494 26.0125
Al 29.3538 32.2176 23.9875
Al 30.7857 33.6494 21.9625
Al 32.2176 0.7159 28.0375
Al 33.6494 2.1478 26.0125
Al 32.2176 0.7159 23.9875
Al 33.6494 2.1478 21.9625
Al 32.2176 3.5797 28.0375
Al 33.6494 5.0116 26.0125
Al 32.2176 3.5797 23.9875
Al 33.6494 5.0116 21.9625
Al 32.2176 6.4435 28.0375
Al 33.6494 7.8754 26.0125
Al 32.2176 6.4435 23.9875
Al 33.6494 7.8754 21.9625
Al 32.2176 9.3073 28.0375
Al 33.6494 10.7392 26.0125
Al 32.2176 9.3073 23.9875
Al 33.6494 10.7392 21.9625
Al 32.2176 12.1711 28.0375
Al 33.6494 13.6030 26.0125
Al 32.2176 12.1711 23.9875
Al 33.6494 13.6030 21.9625
Al 32.2176 15.0349 28.0375
Al 33.6494 16.4667 26.0125
Al 32.2176 15.0349 23.9875
Al 33.6494 16.4667 21.9625
Al 32.2176 17.8986 28.0375
Al 33.6494 19.3305 26.0125
Al 32.2176 17.8986 23.9875
Al 33.6494 19.3305 21.9625
Al 32.2176 20.7624 28.0375
Al 33.6494 22.1943 26.0125
Al 32.2176 20.7624 23.9875
Al 33.6494 22.1943 21.9625
Al 32.2176 23.6262 28.0375
Al 33.6494 25.0581 26.0125
Al 32.2176 23.6262 23.9875
Al 33.6494 25.0581 21.9625
Al 32.2176 26.4900 28.0375
Al 33.6494 27.9219 26.0125
Al 32.2176 26.4900 23.9875
Al 33.6494 27.9219 21.9625
Al 32.2176 29.3538 28.0375
Al 33.6494 30.7857 26.0125
Al 32.2176 29.3538 23.9875
Al 33.6494 30.7857 21.9625
End
Lattice
34.36538957 0.0 0.0
0.0 34.36538957 0.0
0.0 0.0 50.0
End
End
Bulk aluminum has an fcc crystal structure, with a lattice constant of 4.05 Å of the conventional unit cell.
To create the surface, we first build the primitive unit cell of bulk aluminum:
In your molecule editor screen you should see a picture of the bulk aluminum structure. The primitive unit cell contains one atom. As repeated cells are not shown by default, it had to be turned on explicitly. Otherwise you see only the single atom in the unit cell.
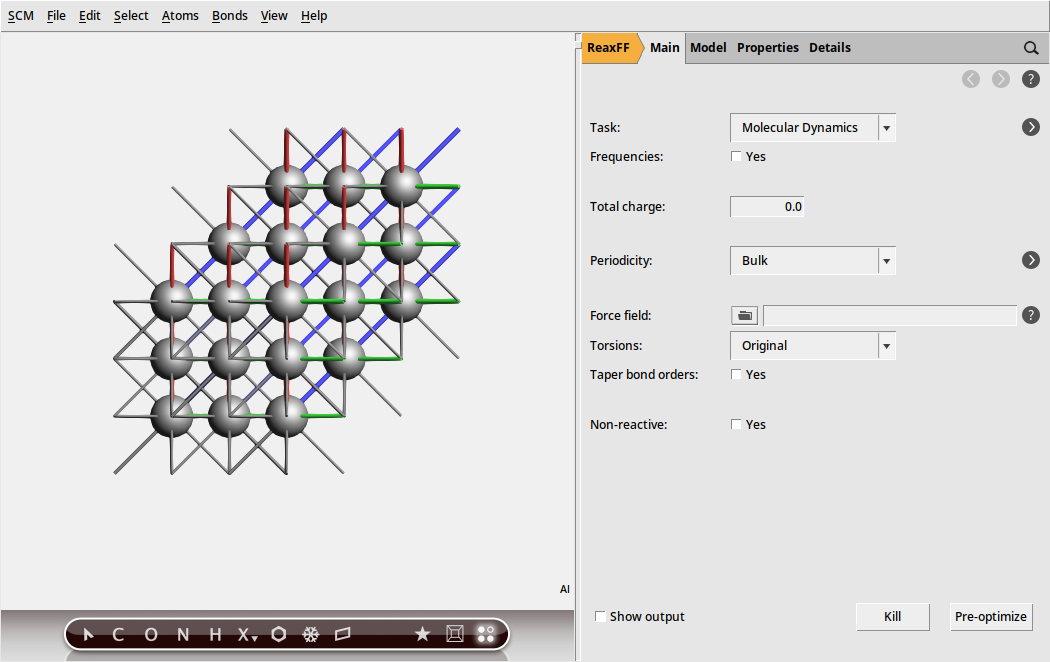
Tip
In the menu bar, select Properties → Properties (including Estimated) to see some properties of the system, such as stoichiometry, angles between lattice vectors, and the system density.
Close the properties window (if not already closed) to return to the ReaxFF window
Surfaces are constructed by specifying the Miller indices, which requires that the conventional (and not primitive) cell be used.
Now create the surface:
0, 0, 1.2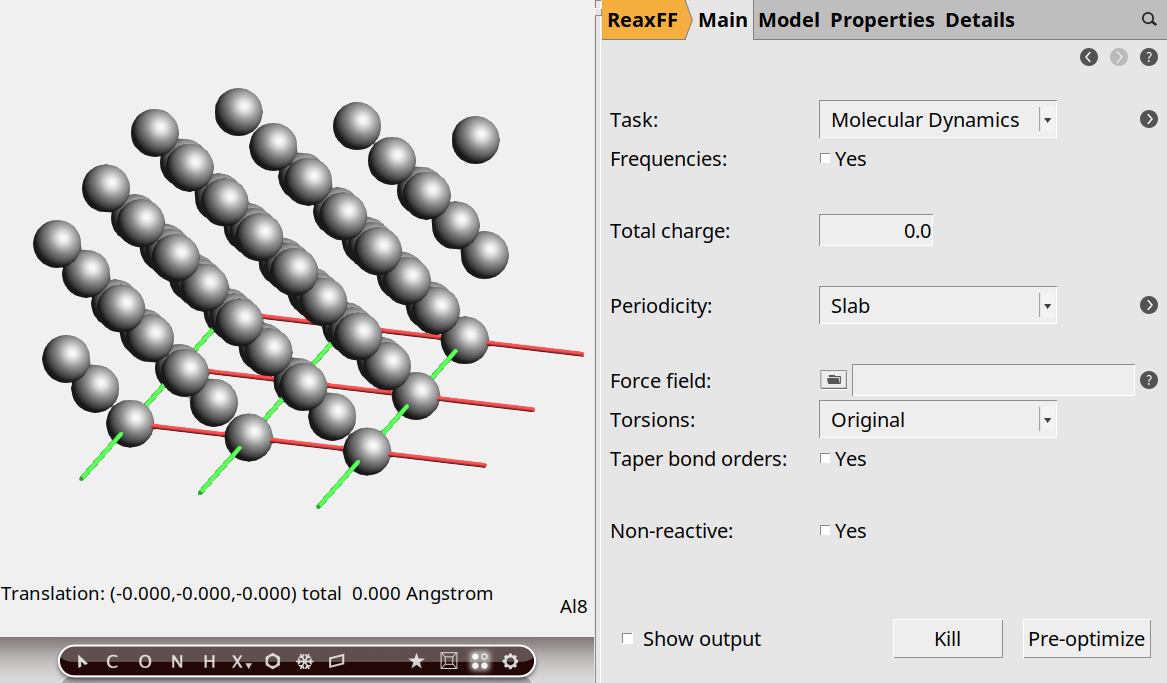
You should be able to see it is a four-layer slab.
In this case we do not want just one unit cell, but a much bigger piece of the slab. So let’s create a 9x9 supercell:
9, 9 on the diagonal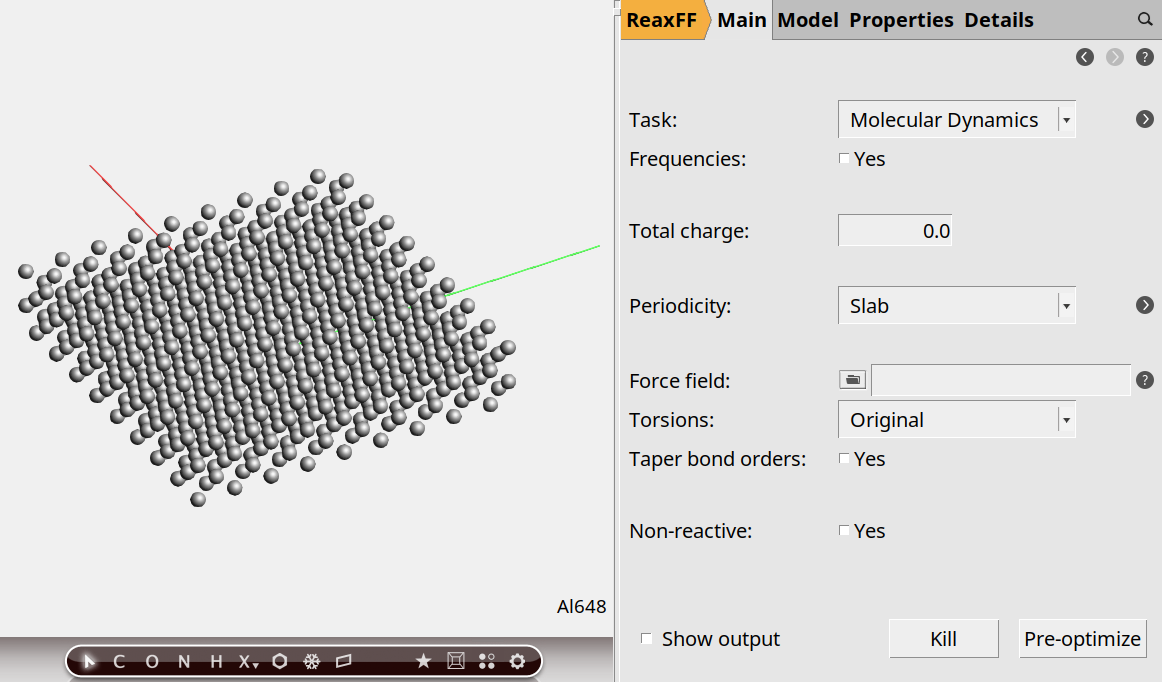
Now we have a slab of aluminum, four layers thick.
When you create a slab in ReaxFF, the periodicity is automatically set to Slab. This means 2D-periodic boundary conditions.
For the solid-liquid interface, we want 3D-periodic boundary conditions:
50 angstrom:To help with visualization, it is often convenient to center the slab in the box:
This moves the Al atoms to the center of the box.
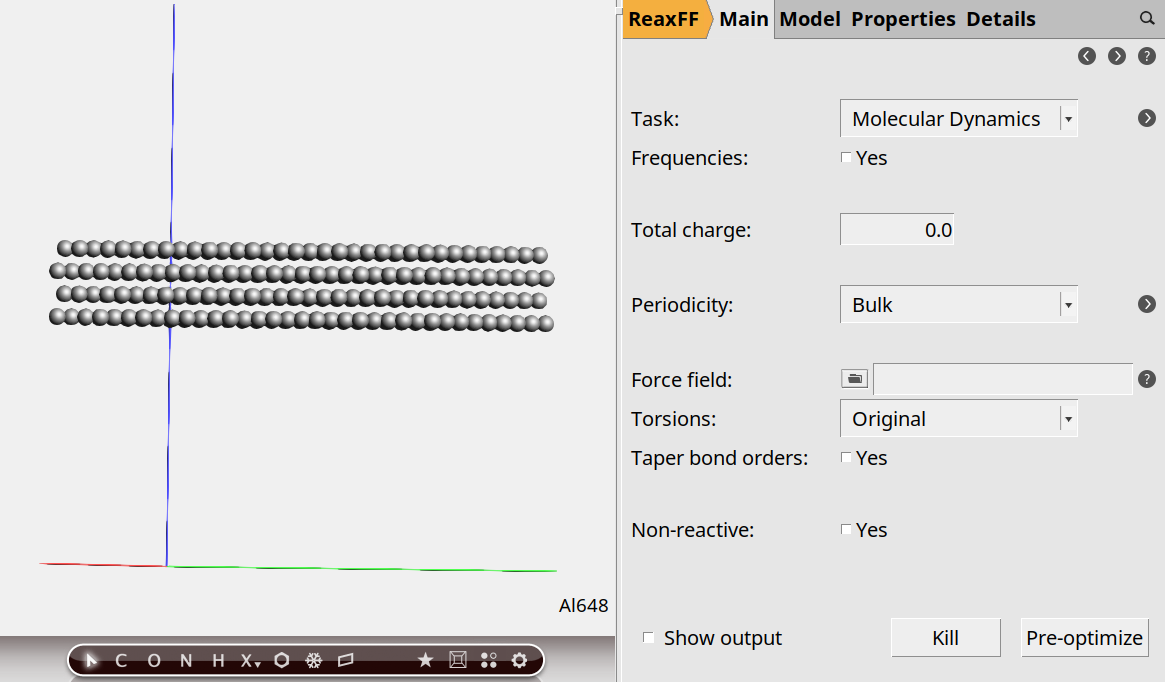
Step 3: Add solvent¶
The next step is to add water to the system.
When the system is 3D-periodic like in this case, the Builder tool will pack molecules around the already existing system.
Note
The Builder interface changed in AMS2025.
In this tutorial we will heat the water quite a lot, so we will use a lower density of 0.72 g/cm³.
0.72 g/cm³.The density refers to the density of the added water molecules, excluding the Al slab. Note that the density specified in this way is approximate, since an estimate is made for the free volume to pack the water molecules in.
OWater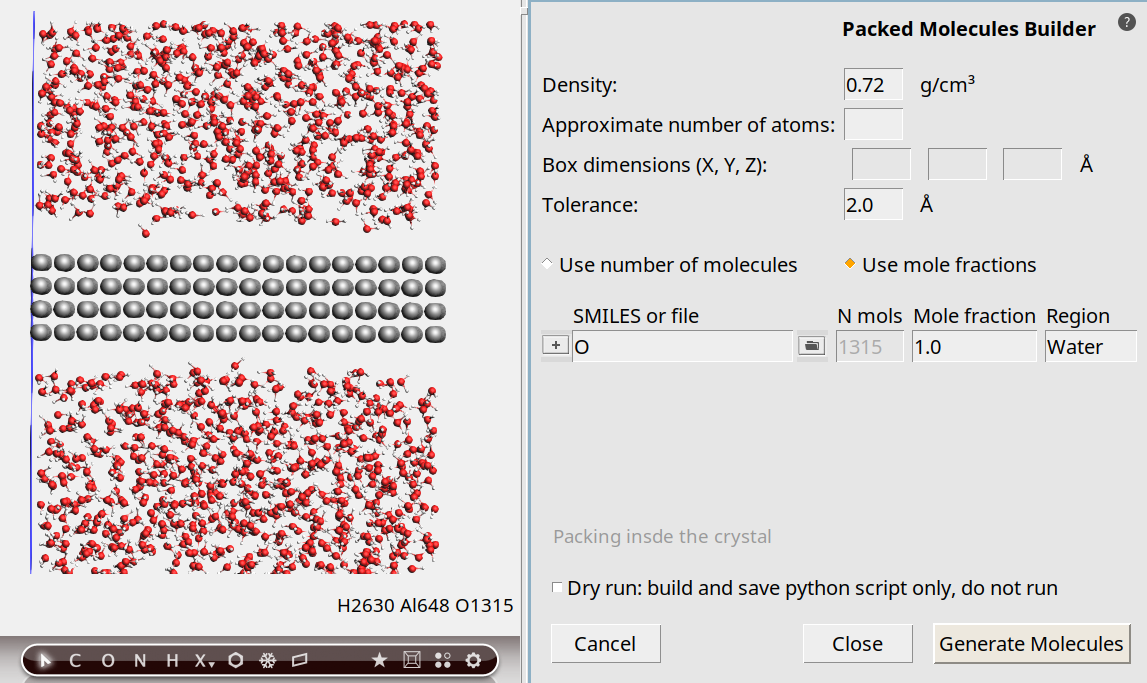
Step 4: Set up the simulation, including a temperature regime¶
Now we will set up the MD-simulation.
For the purpose of this tutorial we want to quickly see something happen in our simulation. We will therefore use a very high temperature for the water, but try to keep the aluminum cool. Also, we will start with a low temperature MD to relax the initial set-up. This can all be accomplished using several thermostats for different regions.
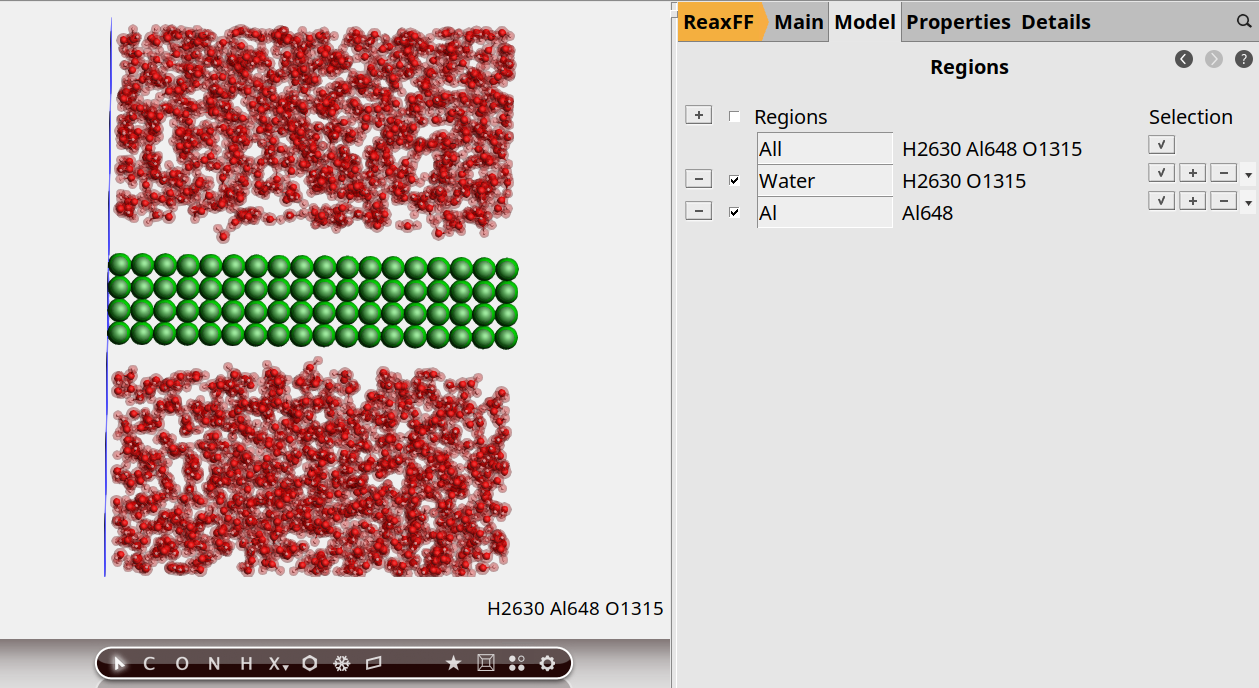
For this we first need to define two regions: one for the aluminum slab, and one for the water. In AMSinput, regions are just defined as a collection of atoms, which can be set up via the Regions panel:
This shows the previously defined water region.
Let us also make a region that contains just the aluminum slab.
Select → Select atoms of Same… TypeRegion_2 to AlWater region to highlight itYou should now see the two different regions highlighted in different colors:
We will use the Al-water force field and a Nose-Hoover thermostat with a default damping constant of 100 fs:
 to go the Model → MD panel
to go the Model → MD panel1000000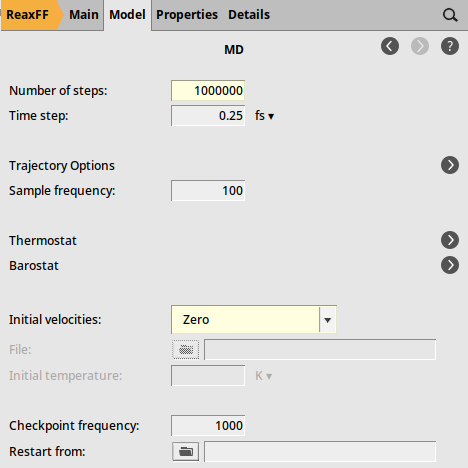
By default, the AMS MD driver records a lot of data for potential further analysis. This can consume considerable amounts of disk space for long MD simulations (like the one million steps we perform here). It might often be worth customizing the trajectory sampling settings to reduce the amount of data saved if you are sure you will not need them afterwards. In our case, we know we will not perform any analysis based on atomic velocities, and we also do not need any of the engine-specific data stored on engine results files (MDStep*.rkf). For ReaxFF, these files contain detailed bonding tables including very weak bonds, as well as a breakdown of individual potential energy terms.
next to Trajectory Options0
Now that we have defined the regions we needed, we will set up the temperature regime. We will start the water with a temperature of 300 K for 4000 steps, then warm it up to 2000 K within 4000 steps and maintain this temperature for the rest of the simulation.
next to Thermostat100.0 fs300 300 2000 into the Temperature(s) field4000 4000 into the Duration(s) fieldHint
Note that with time-dependent thermostats the set temperatures are always connected with a linear ramp. The Temperatures field should therefore always contain one number more than the Durations field, as the last temperature will be held indefinitely. In the example above, in order to have a constant temperature of 300 K in the beginning, we explicitly specified that the temperature should go from 300 K to 300 K within the first 4000 steps.
We now have the time-dependent thermostat of the water set up. Let us add another thermostat that just keeps the aluminum slab cold.
300 K100.0 fsYour thermostat setup should look like this:
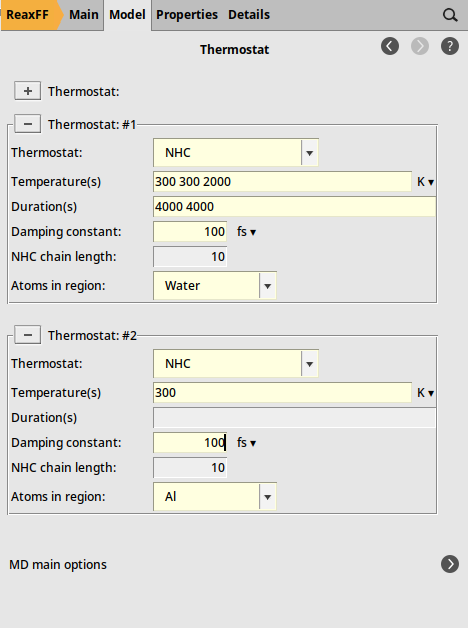
Step 5: Run the simulation¶
Now we can run our set up:
Al-water as filename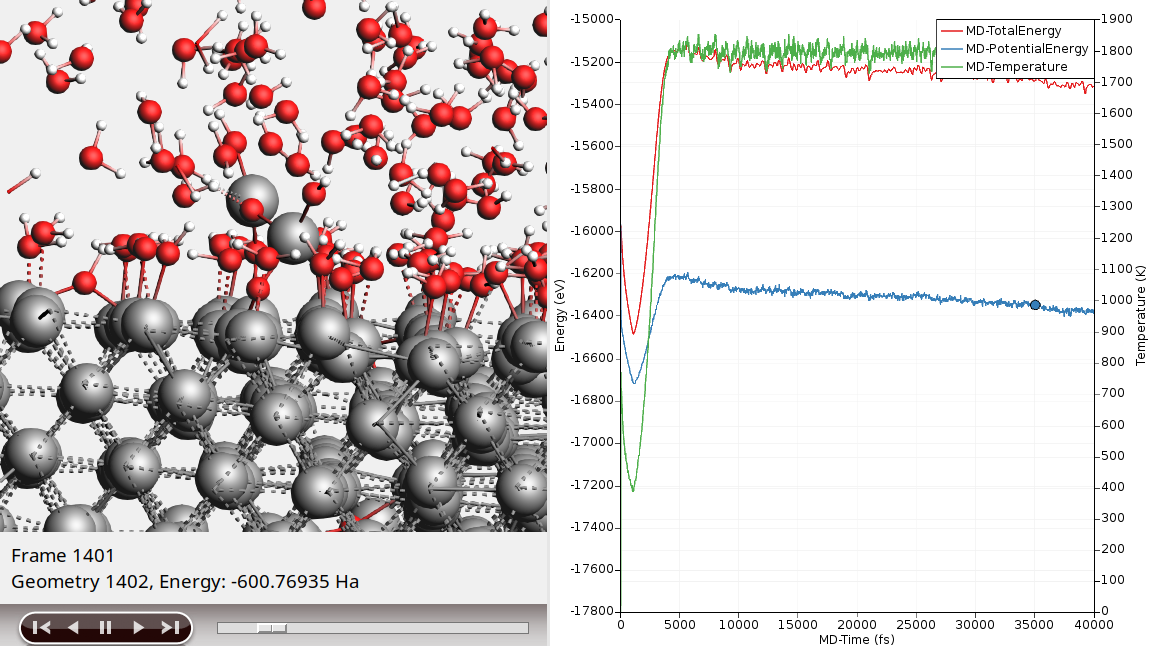
You will see many water molecules adsorb on the aluminum surface almost immediately. Around frame 40 (that is MD step 4000) you will see the temperature increase, as the water thermostat starts heating its region. After some time, you will also see some of the adsorbed water molecules lose one hydrogen. The lone proton will likely drift off into the water, while the remaining OH goes into a bridge-like configuration with two of the surface Al. This configuration can already be seen in the picture above.
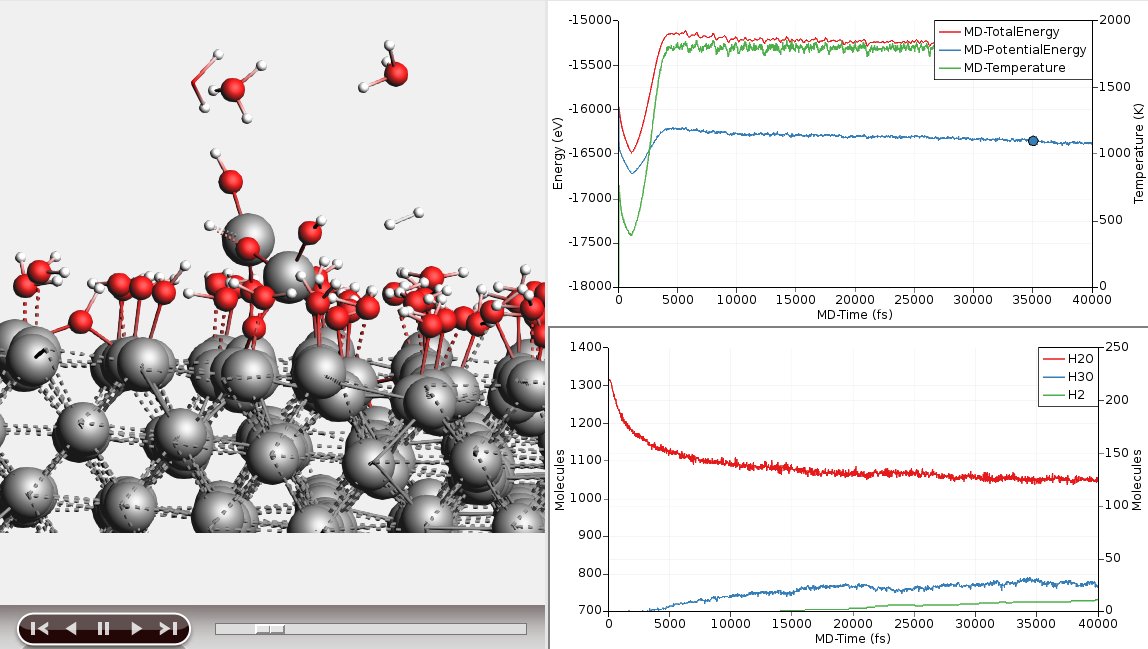
You can leave the simulation running to see what will happen. It will take a long time though. If you do leave it running for a while, you will see that the OH in the aforementioned bridge configuration tend to pull one of the Al atoms out of the surface, essentially roughening it and allowing more water to adsorb. You might also see aluminum atoms completely detach from the surface an become dissolved in the water layer as AlO3H3. Eventually a rough aluminum oxide layer forms, while the split off protons combine to form molecular hydrogen.
You can download the movie here if it does not play in your browser.
We can use the molecular composition analysis tools in AMSmovie to get a bit clearer picture of that process. Let us plot the number of H2O, H3O and H2 molecules in our simulation:
If you do not want to wait for the simulation to finish, you can now request the job to stop:
Your job will quit after the next sampled MD step.