Glass transition temperature¶
In this ReaxFF tutorial the calculation of glass transition temperatures, Tg, from the temperature dependence of the polymer’s density is explained.
The systems and workflows presented here are originally described in the publication Effect of chemical structure on thermo-mechanical properties of epoxy polymers: Comparison of accelerated ReaxFF simulations and experiments, Polymer 159, 354-368 (2018).
The tutorial consists of the following steps:
Note
The calculations are computationally demanding. For optimal performance, a parallel execution on a compute cluster is advised. This can best be done by using the remote job management of the GUI
Importing the polymer structure¶
The tutorial will use a cross-linked epoxy polymer generated with the bond boost method (see also bond boost tutorial ).
Begin by downloading the polymer and import into AMSinput:
here to download the .xyz file detda-epoxy.xyz
Simulated annealing¶
In order to accurately simulate the temperature dependence of the density, we will have to make sure that the polymer structure is as homogeneous as possible. Large holes inside the polymer structure are an indicator for an inhomogeneous structure that will result in very noisy and hard to analyze results.
Simulated annealing is a simple optimization strategy intended to yield the global minimum energy structure on a complex and high dimensional potential energy surface. During the annealing the temperature during an MD simulation is gradually increased, hoping that the system can overcome more and more energy barriers and explore the energy landscape effectively. The heating phase is followed by a cool down period in which the system is cooled back down to the initial temperature. Often the resulting structure is significantly lower in energy than the starting structure, though there is no guarantee it is the optimal one.
Start by choosing the MD settings:

 next to Task: Molecular Dynamics to go to the MD details
next to Task: Molecular Dynamics to go to the MD details9700005000
To allow the unit cell to shrink or expand we further add a barostat:
next to Barostat1.0 atm.500 fs.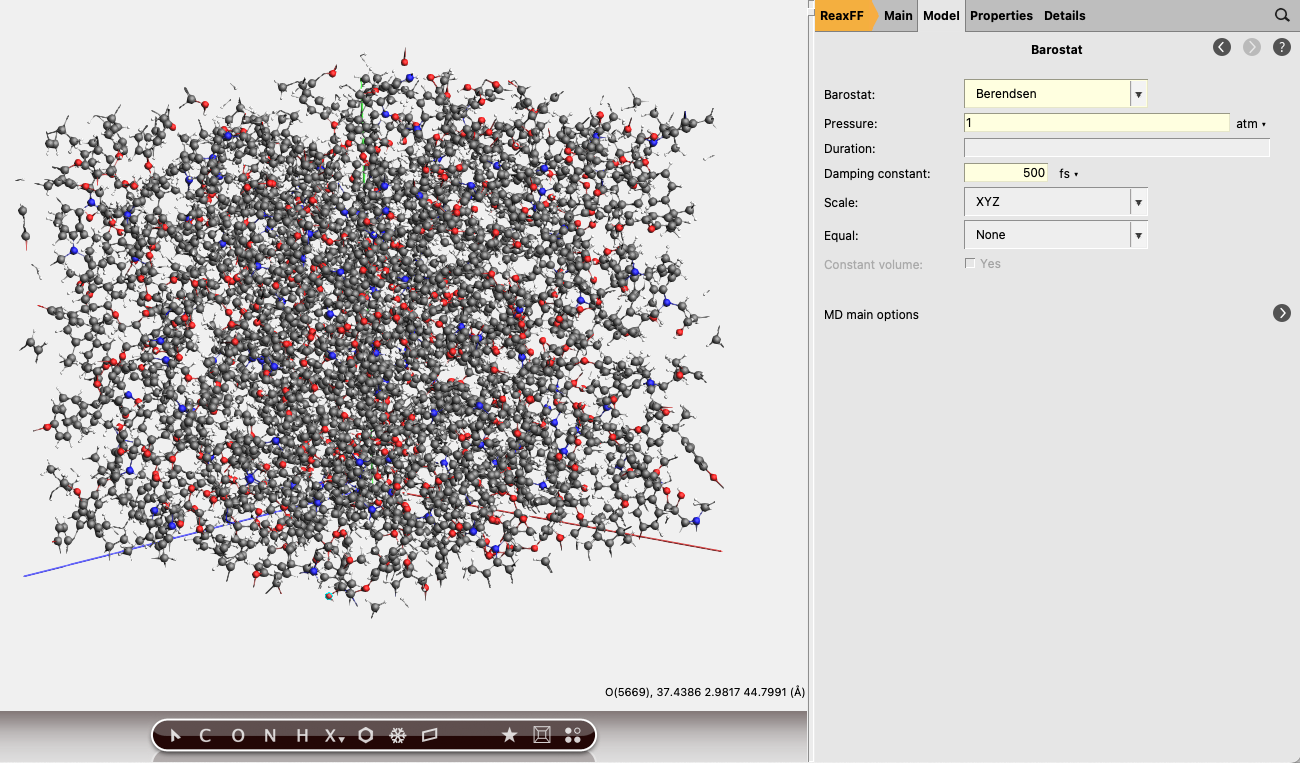
The simulated annealing is defined in the thermostat panel
next to MD Main options next to Thermostat button to add a thermostat to the simulation.
button to add a thermostat to the simulation.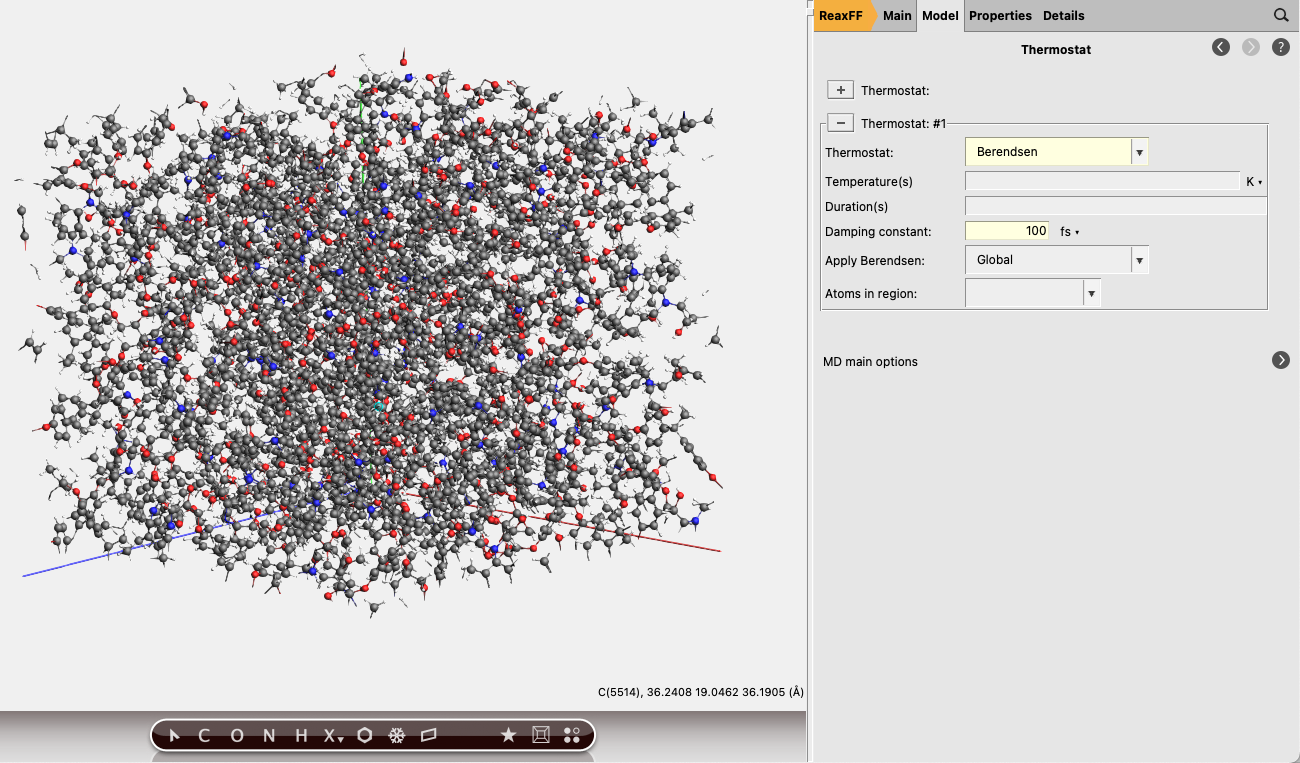
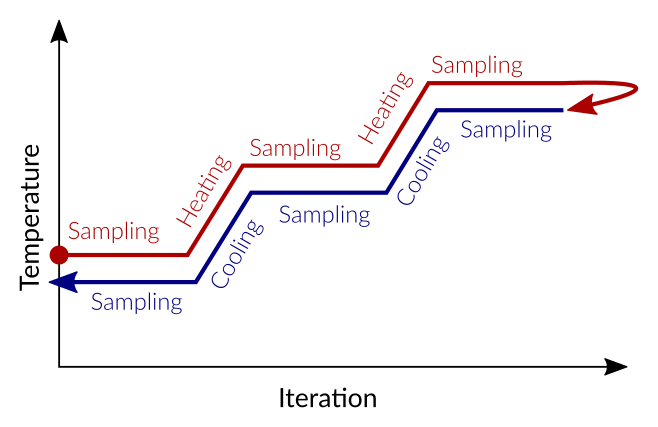
Next, we define the desired temperature program. For the extraction of densities from the calculation we shall increase the temperature from 298.15 to 598.15 which is still low for a simulated annealing but sufficient for our purpose.
The temperature program we want to implement is simple:
Heat up by 25 K over the course of 30000 steps
Sample the density over a course of 10000 steps
Repeat until reaching 598.15 K, then reverse to cool down to 298.15 K again
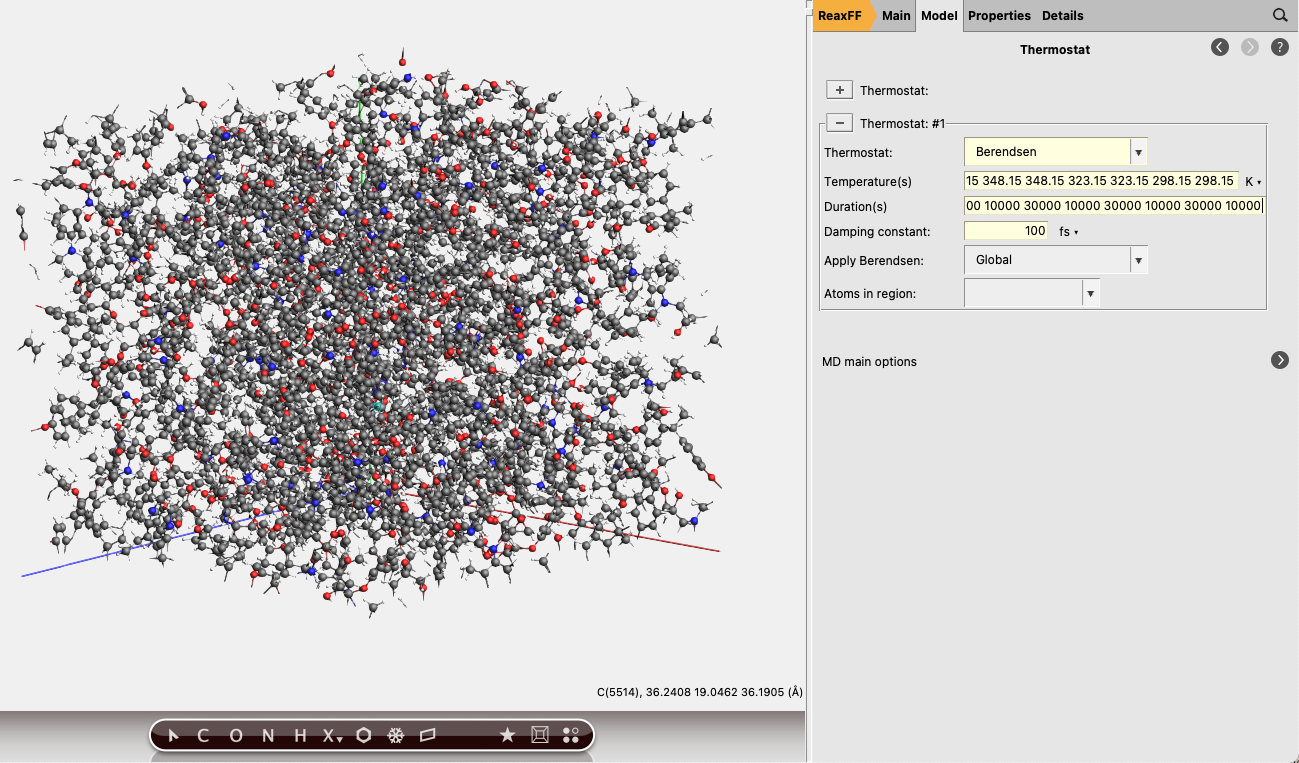
The individual steps of this program can be entered into the input fields Temperature(s) and Durations. In our case it’s easiest to copy and pastem them into the GUI.
Temperatures:
298.15 298.15 323.15 323.15 348.15 348.15 373.15 373.15 398.15 398.15 423.15 423.15 448.15 448.15 473.15 473.15 498.15 498.15 523.15 523.15 548.15 548.15 573.15 573.15 598.15 598.15 573.15 573.15 548.15 548.15 523.15 523.15 498.15 498.15 473.15 473.15 448.15 448.15 423.15 423.15 398.15 398.15 373.15 373.15 348.15 348.15 323.15 323.15 298.15 298.15
Durations:
10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000 30000 10000
We are now ready to start the simulated annealing calculation
Extraction of Density vs. Temperature profiles¶
To extract the densities and temperatures from the trajectory for post-processing we make use of a Python script.
densities.py from hereNext, you need to open the command line to execute the script with the AMS Python interpreter.
Tip
Windows and Mac users should open a pre-configured command-line from the GUI
In the command line, the script can be executed as follows
$AMSBIN/amspython densities.py SimulatedAnnealing.results
This will print a table to the terminal from where it can be copy-pasted, or the data can be written to a file by appending > someFileName.csv to the end of the command above.
Calculation of the glass transition temperature¶
The density profiles can be plotted with any graph plotting software, e.g. gnuplot or LibreOffice calc. Import the data into the plotting software of choice and plot the two columns against each other. The graph will fall into one of two categories:
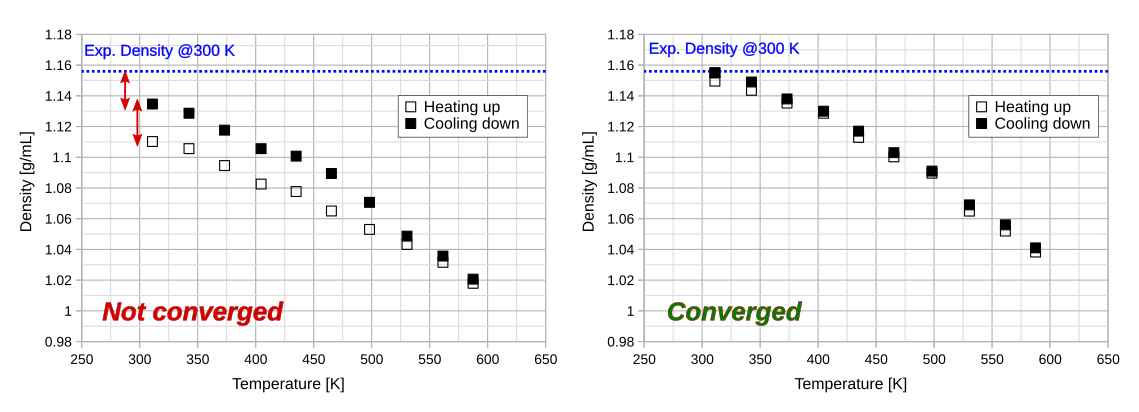
If the final density of the initial structure and the equilibrated structure differ significantly and the experimental density (if available) is far off, then the density is not converged and another simulated annealing run should be carried out. If the final and initial density are close, then the simulation can be considered converged and one can proceed with the calculation of Tg.
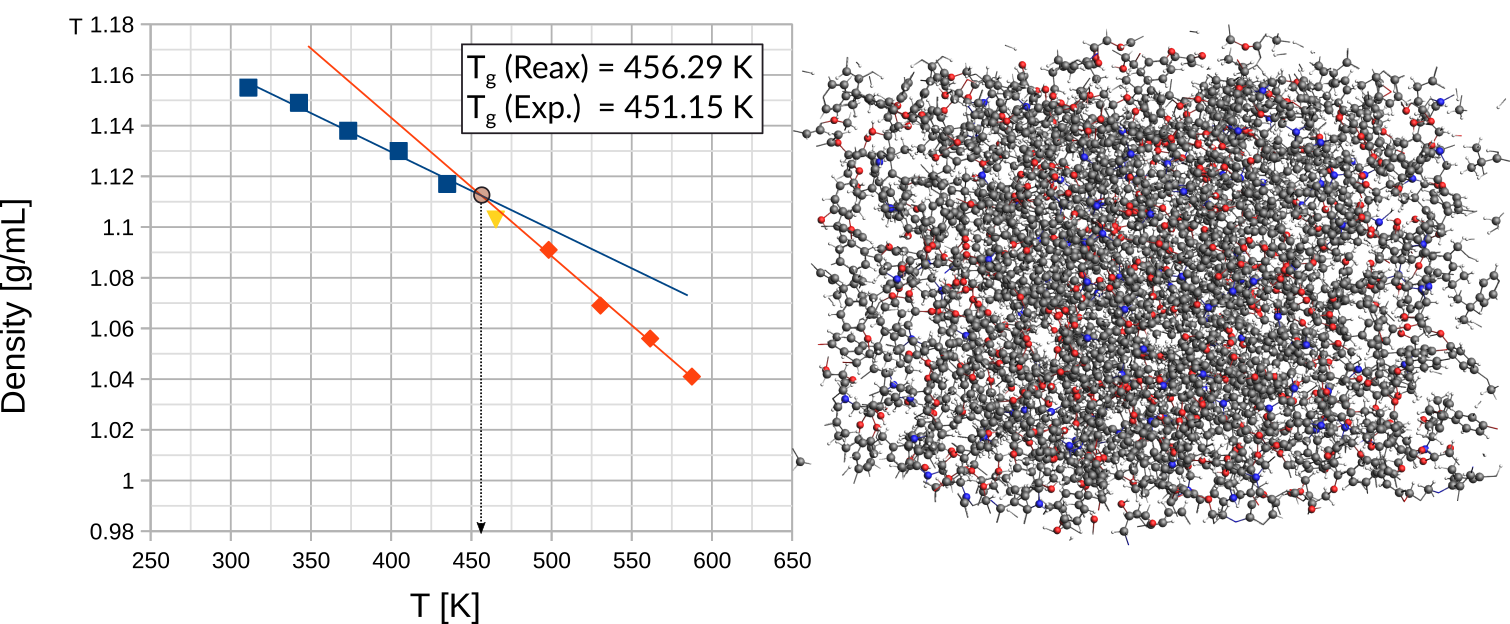
To calculate Tg, carry out two linear fits through the data points as shown in the below figure. The intersection between these two lines marks the glass transition temperature.
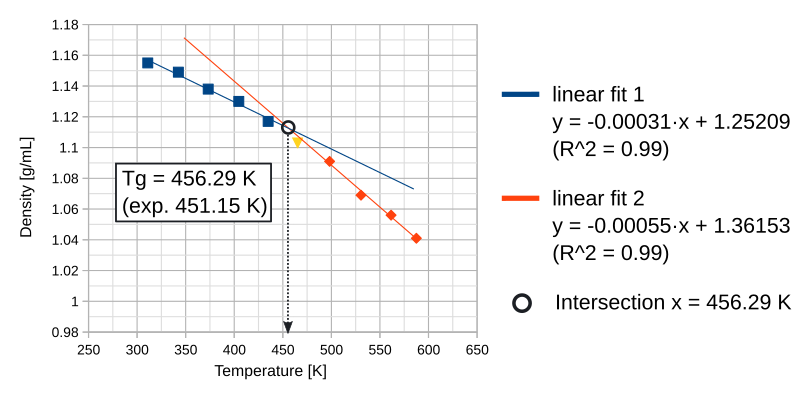
The subsets of points for the linear fits should be chosen such that a maximum R2value is obtained. If the R2values are too low, i.e. the data is too noisy, or the curves don’t intersect, we advise to try one of the following troubleshoots:
Run another simulated annealing simulation
Extend the temperature range
Reduce or increase the averaging parameter in the densities.py script