Combustion simulation (ReaxFF)¶
In this tutorial we will set up a reactive MD simulation using ReaxFF for modeling the combustion of methane gas.
This tutorial will show you how to:
Create a simple mixture (methane and oxygen)
Set up a quick combustion reaction with ReaxFF
Monitor and visualize the combustion process
Analyze the reaction
Step 1: Start the GUI¶
 →
→ 
Step 2: Create a methane / oxygen mixture¶
Next, we will make the methane / oxygen mixture. For full combustion, we need to have at least 2 oxygen molecules per methane molecule. To guarantee proper mixing, we choose to use a slightly leaner mixture with a CH₄:O₂ ratio of 1:2.5.
We will use the system builder in AMSinput to construct our mixture:
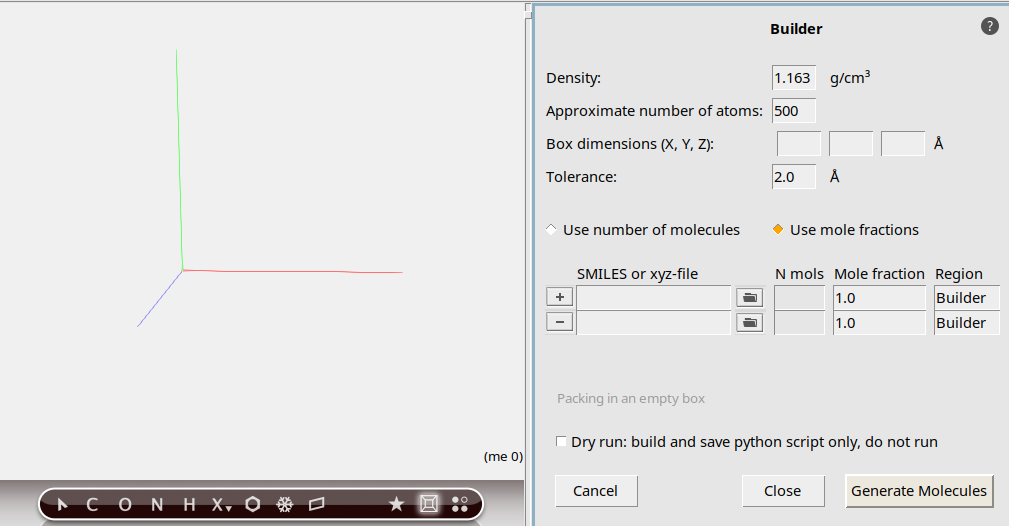
For this tutorial, we will focus on a high compression ratio with a gas density of 1.163 g/mL in order to encourage a fast combustion reaction.
met to search for methane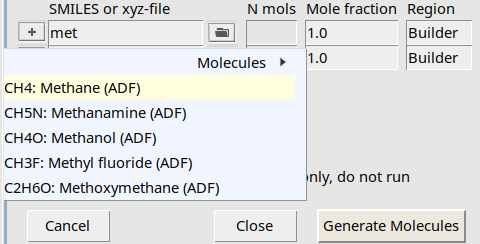
A list with search suggestions will appear, similar to the search box ( ) in the panel bar.
) in the panel bar.
o to search for O2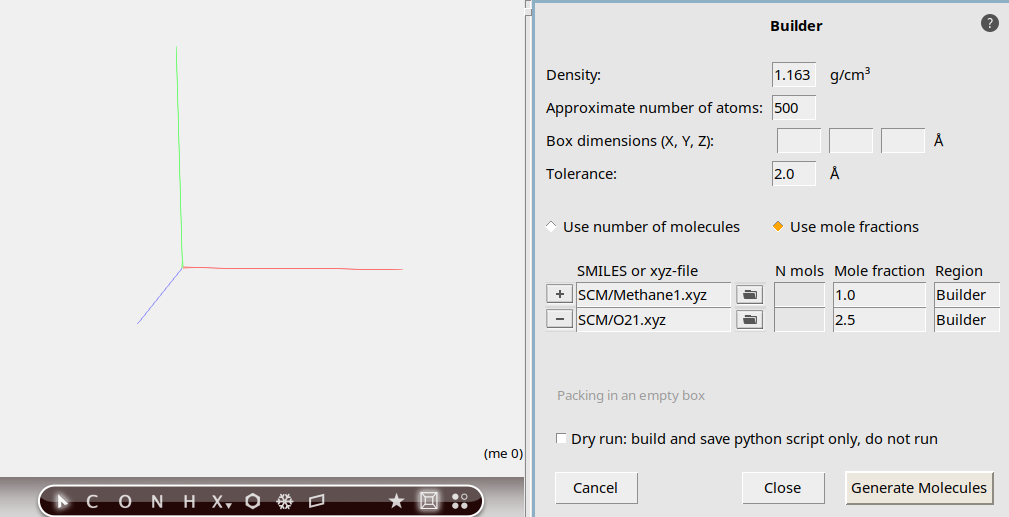
We will now have the builder generate the molecules:
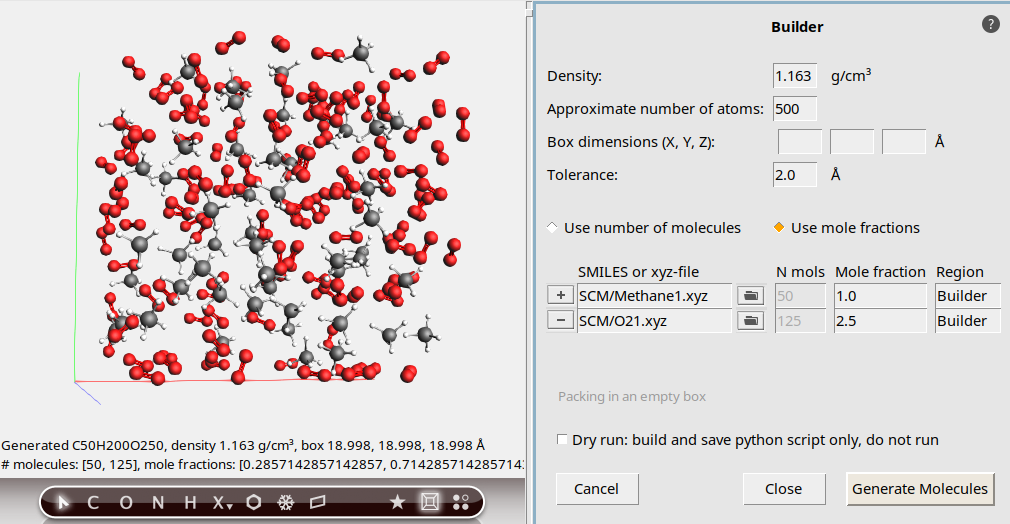
The molecules are generated at random non-overlapping positions and orientations. The output summary tells us the distribution of molecules in our mixture: 50 units of CH₄ and 125 units of O₂. This matches our description of the system, and we can now close the Builder menu.
Step 3: Set up the simulation¶
The next step is to set up the details of the simulation.
Note
For this tutorial, we will perform an MD simulation at both high temperature and density in order to make the combustion process proceed quickly. Typical scenarios using lower temperatures and/or densities will require longer simulation times in order to observe substantial conversion of reactants.
 next to the Force field entry in AMSinput
next to the Force field entry in AMSinputThis will open a new window with a list of available force fields, including a short description of the relevant systems and references. For this particular example we will use the CHO force field for hydrocarbon oxidation.
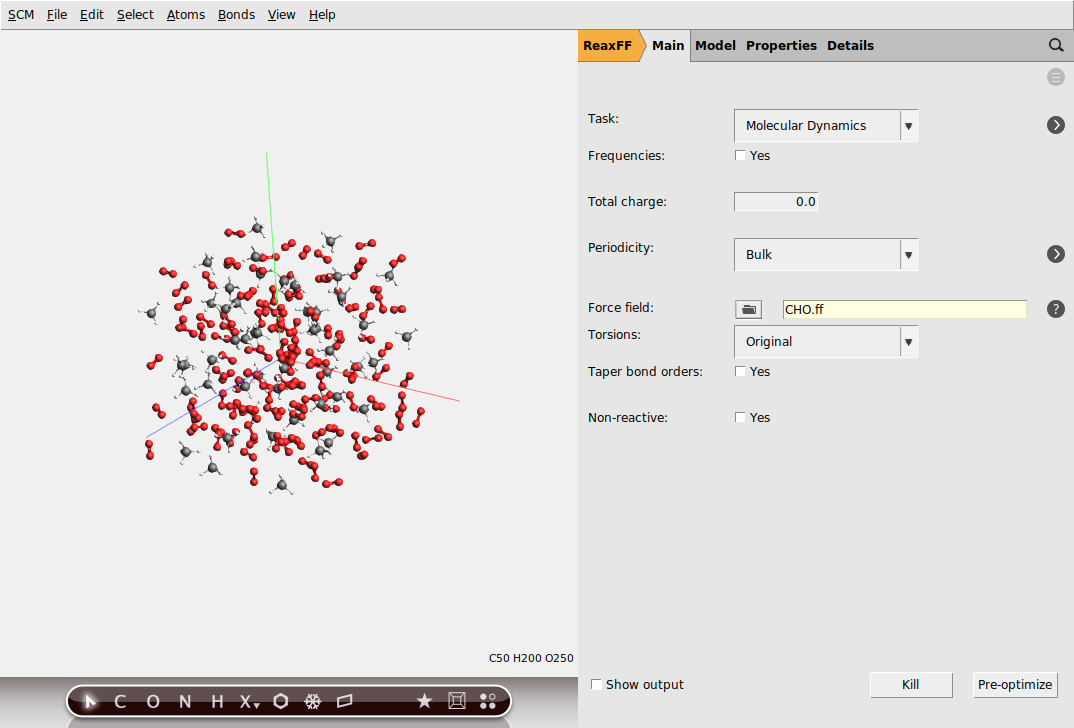
Next, we will configure the MD settings:
 next to Molecular Dynamics
next to Molecular Dynamics30000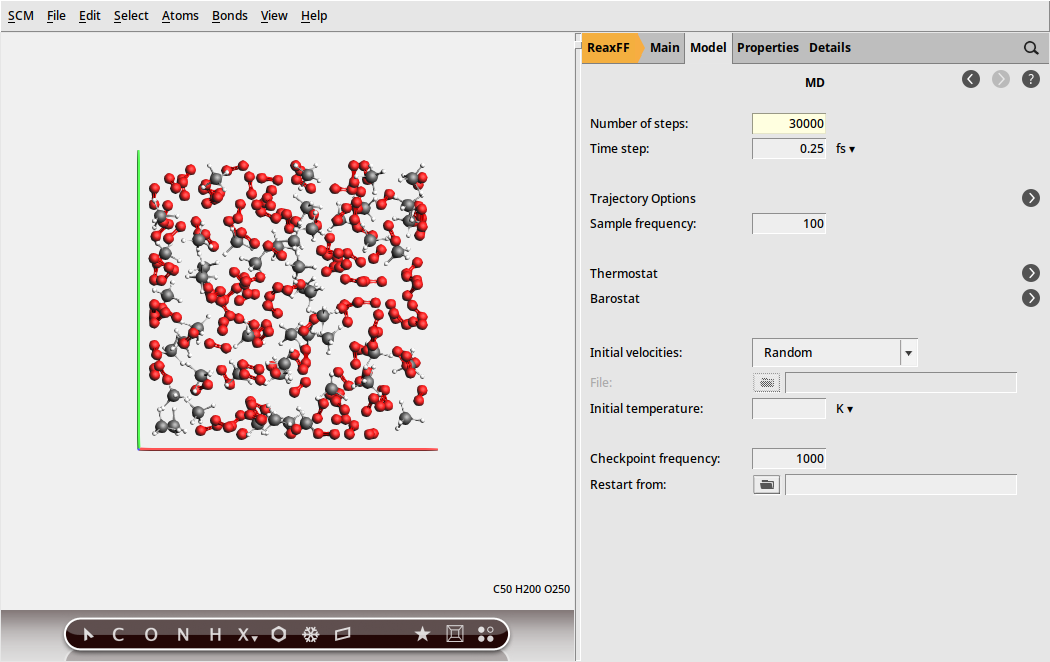
Note
In order to model a constant-temperature system (NVT), we apply a thermostat to the MD simulation. Because energy fluctuations may occur for reactive systems, we choose to use the Nose-Hoover thermostat.
The Nose-Hoover damping constant is dependent on the system size (as it effectively dampens the internal vibrations). For the current system, we use the default value of 100 fs.
When applying the combustion workflow to a realistic system, it may be worth testing different values to see which one yields the best performance.
To configure the Nose-Hoover thermostat:
next to Thermostat button to add a new thermostat
button to add a new thermostat3500 K and a Damping constant of 100 fs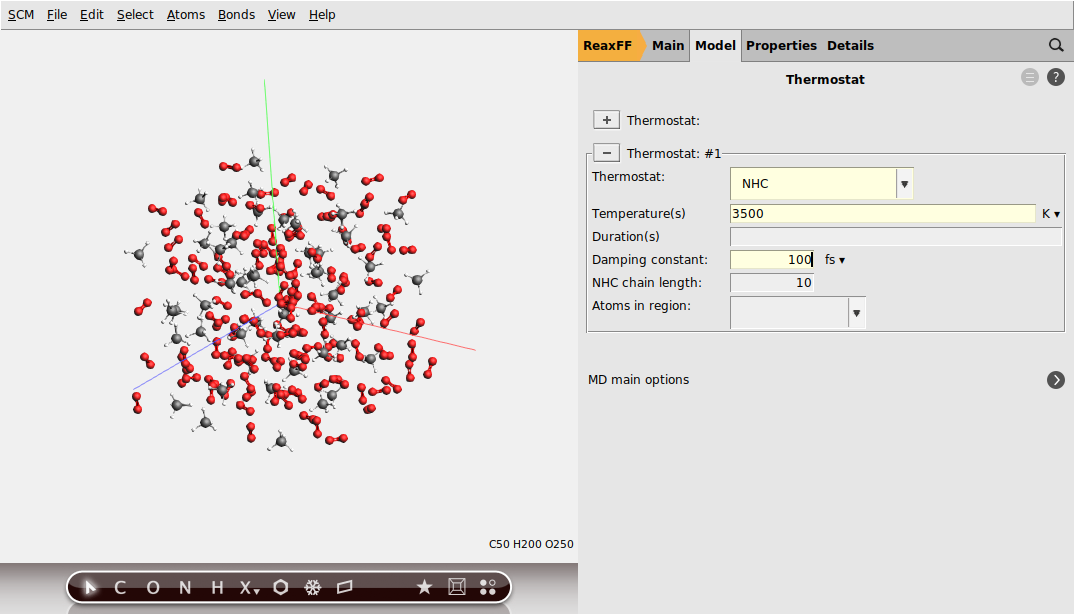
Step 4: Run the simulation¶
We will now run the combustion simulation.
MethaneNote
The simulation will take approximately 10 minutes. You can already proceed with the following steps while the job is running.
AMSjobs will come to the foreground, with the Methane job being shown at the top. Once the simulation starts, progress reports will be shown underneath the active job. We can also monitor the job output while the simulation is running:
Methane job in AMSmovie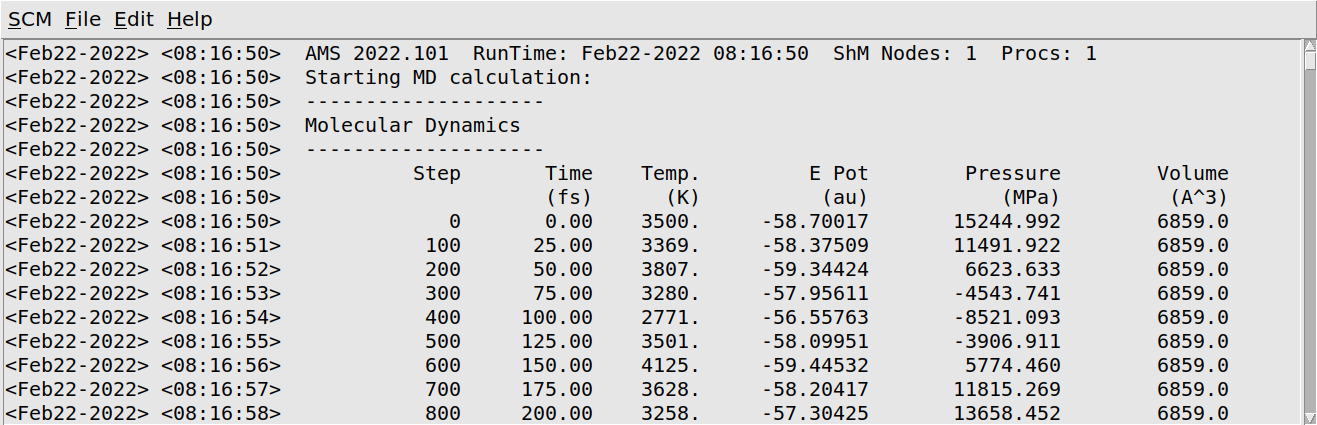
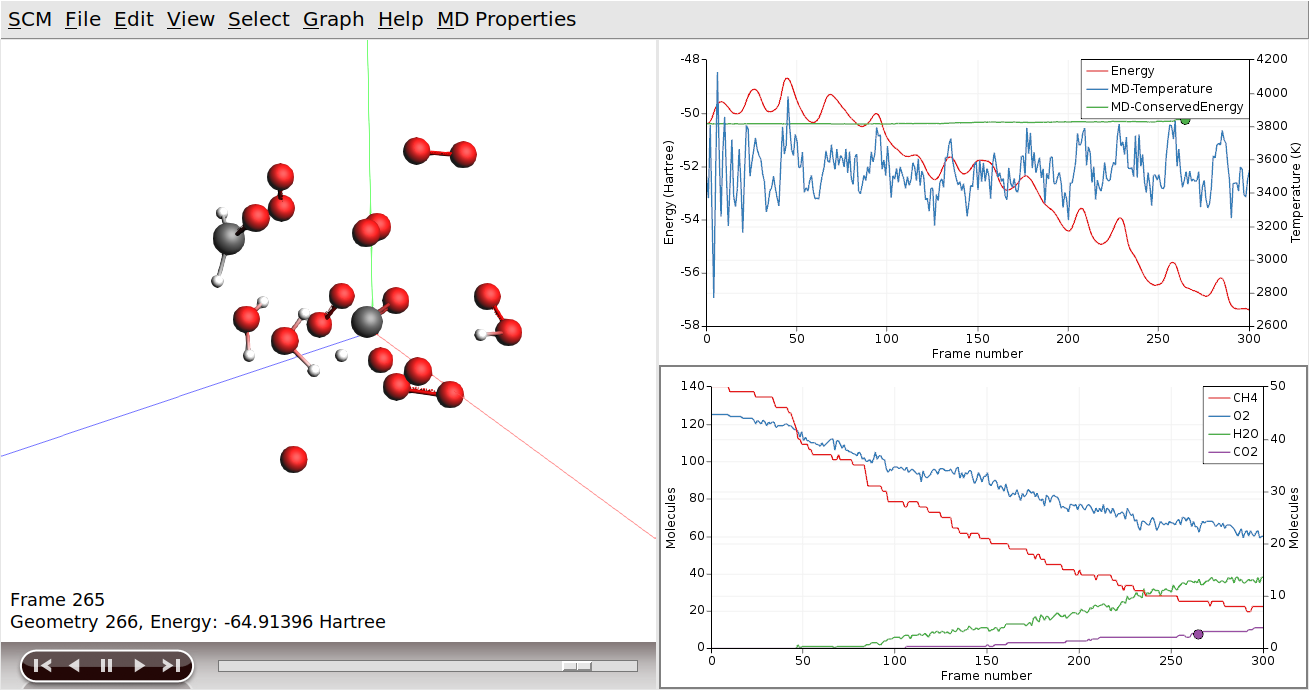
To see how the reaction progresses, we can visualize the output using AMSmovie. This works even while the simulation is still running:
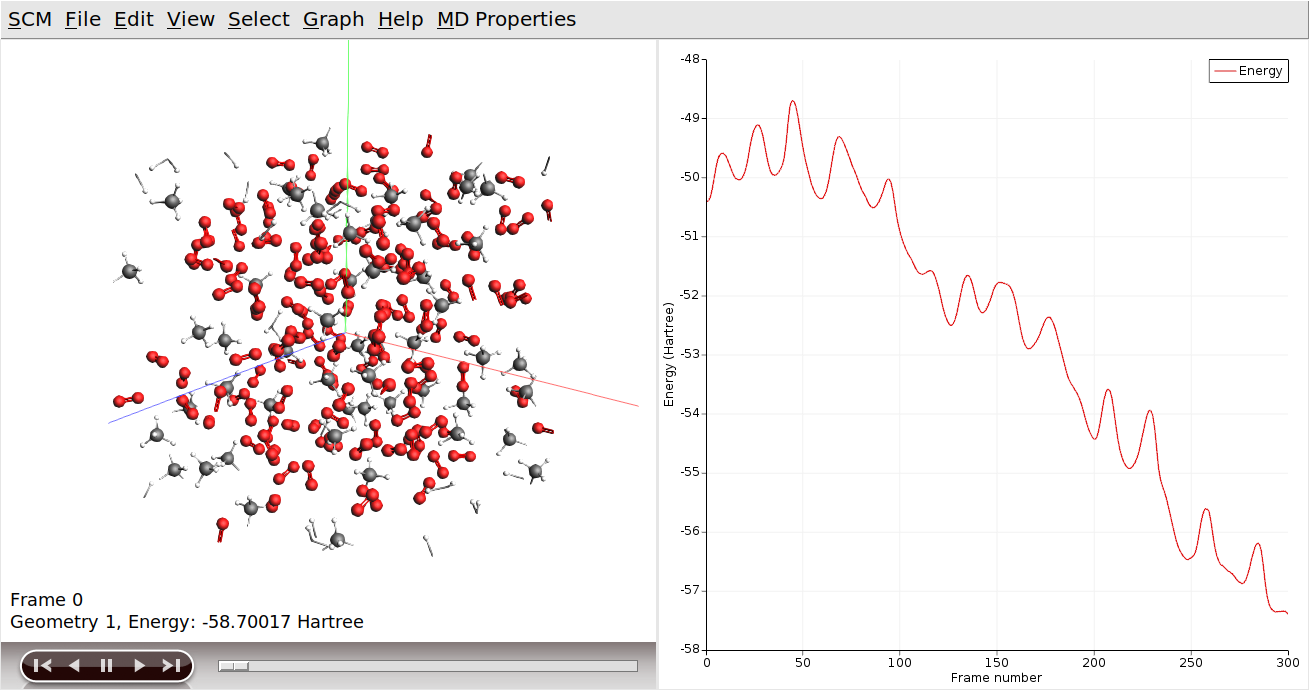
AMSmovie will show the molecular coordinates along the MD trajectory. New data is read in automatically as soon as it becomes available.
AMSmovie can also show graphs with some of the system properties that are calculated by ReaxFF:
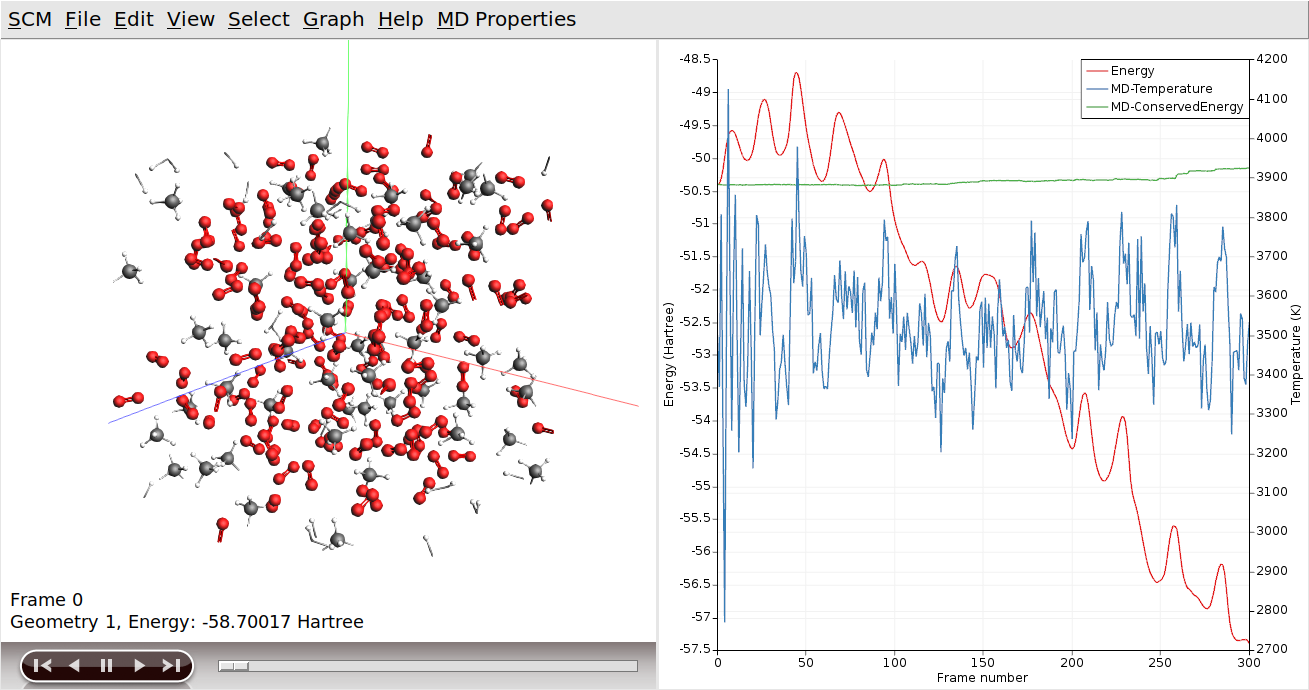
The slider at the bottom of the AMSmovie window can be used to move between different snapshots of the system. You can also click on a specific point in the graphs to pull up the corresponding state. The arrow keys (left and right) can then be used to move through the simulation one image at a time.
Tip
Jump to the end of the movie to automatically monitor the progress of the simulation
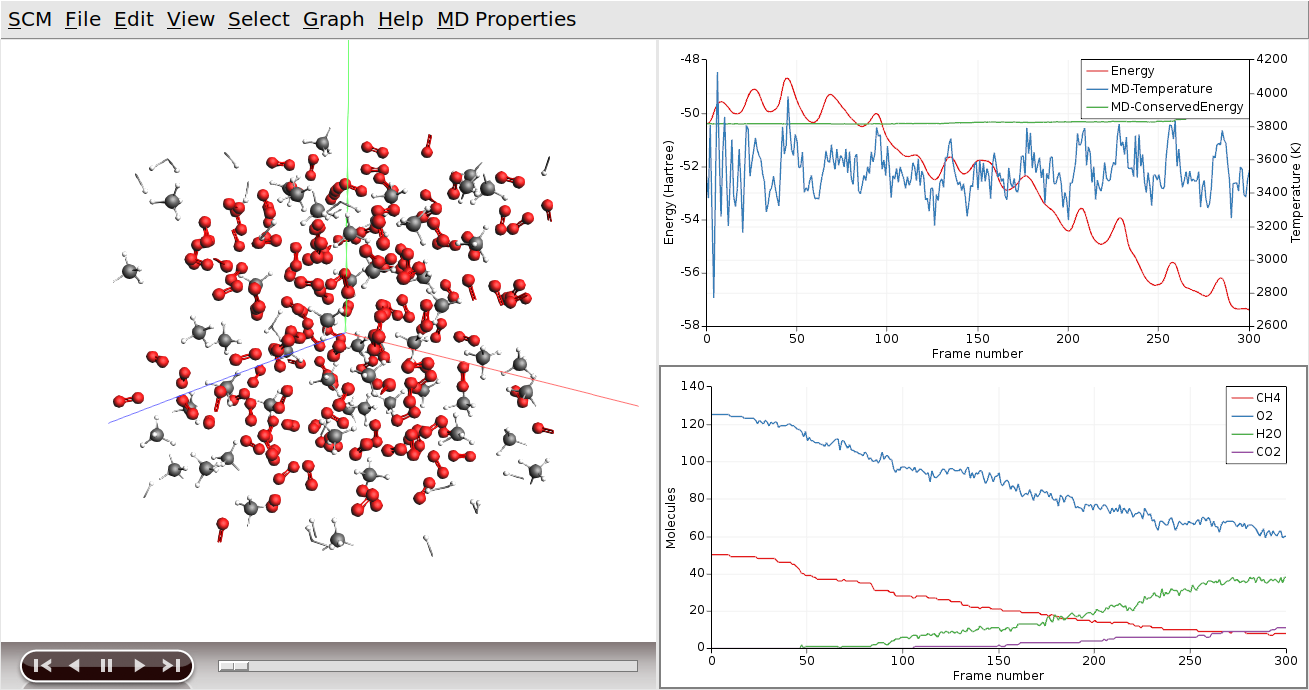
ReaxFF is a reactive force field, which allows reactions to take place during the MD simulation. In our example, methane and oxygen should react to form water and carbon dioxide. We can add graphs to show how many of the different molecule types are present at a given time:
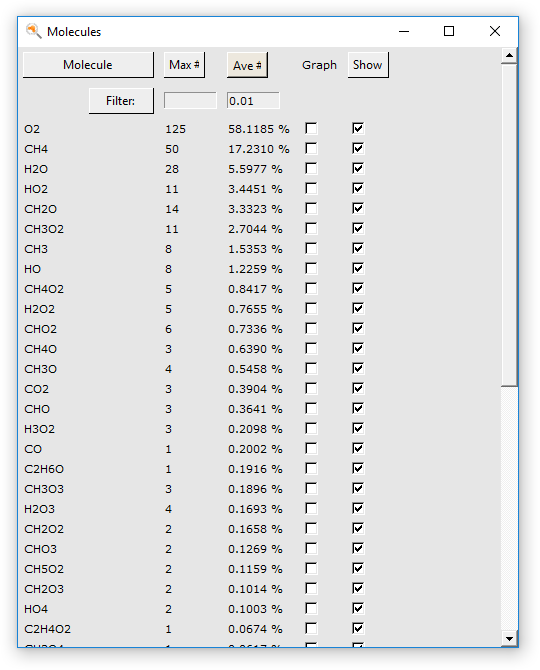
A dialog window will open, showing a list of the molecular species encountered during the simulation. The buttons at the top of the dialog allow sorting the molecules by name, occurrence in the last frame, or average occurrence.
Note
It may take a while before products are formed during the combustion simulation. Some reaction species may not appear in the Molecules menu if the simulation has not progressed far enough.
If this is the case here, you may need to wait a bit longer before new results come in. (Or in some cases, increase the total number of simulation steps.)
Note
The information shown in the Molecules dialog does not update automatically while the simulation is running.
You can refresh the list by closing and re-opening the dialog.
The first check box, labeled “Graph” is used to toggle the graphs for each of the molecular species. These graphs show how the number of molecules changes over time.
The second check box, labeled “Show”, allows you to hide molecules in the 3D model. This is convenient for e.g. monitoring product formation, or focusing on the distribution of specific reactants, as the other molecules can temporarily be hidden from view.
Two filter options are also included. These options hide species from the list for which either the number of occurrences or the mole fraction falls below the specified threshold. This is intended for more complex reaction systems, which can sometimes contain thousands of intermediate species.
Tip
You can use the mouse to move the legends
Sometimes, it can help to put one of the curves on a different axis:
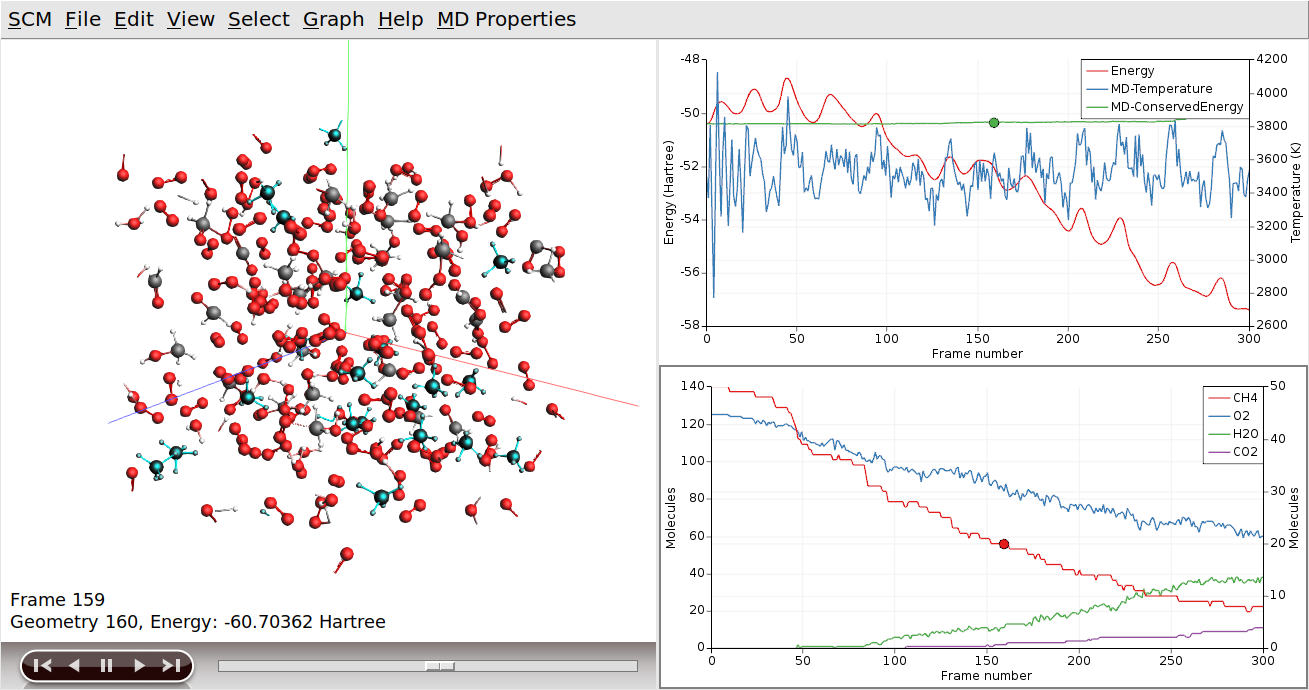
Clicking on the curve also had two other effects (besides making it the active curve): AMSmovie jumped to the snapshot matching the point where you clicked on the curve, and the corresponding molecules (CH4 in this case) are highlighted in the 3D view.
We can use the wireframe view to help make these molecules stand out from the rest:
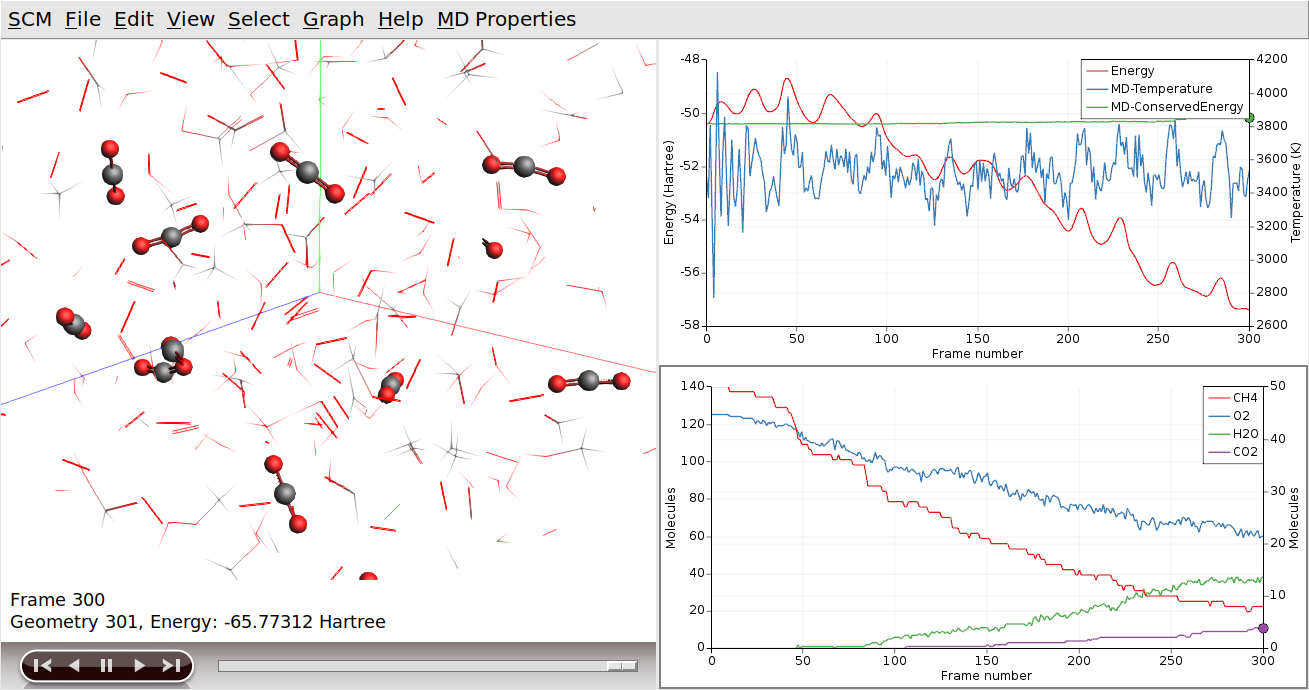
When moving through the movie, it is now easier to see how the reactions actually take place. (Note that atoms will remain selected, even if they are no longer part of a CO2 molecule.)
We can use the same procedure to focus on the H2O production:
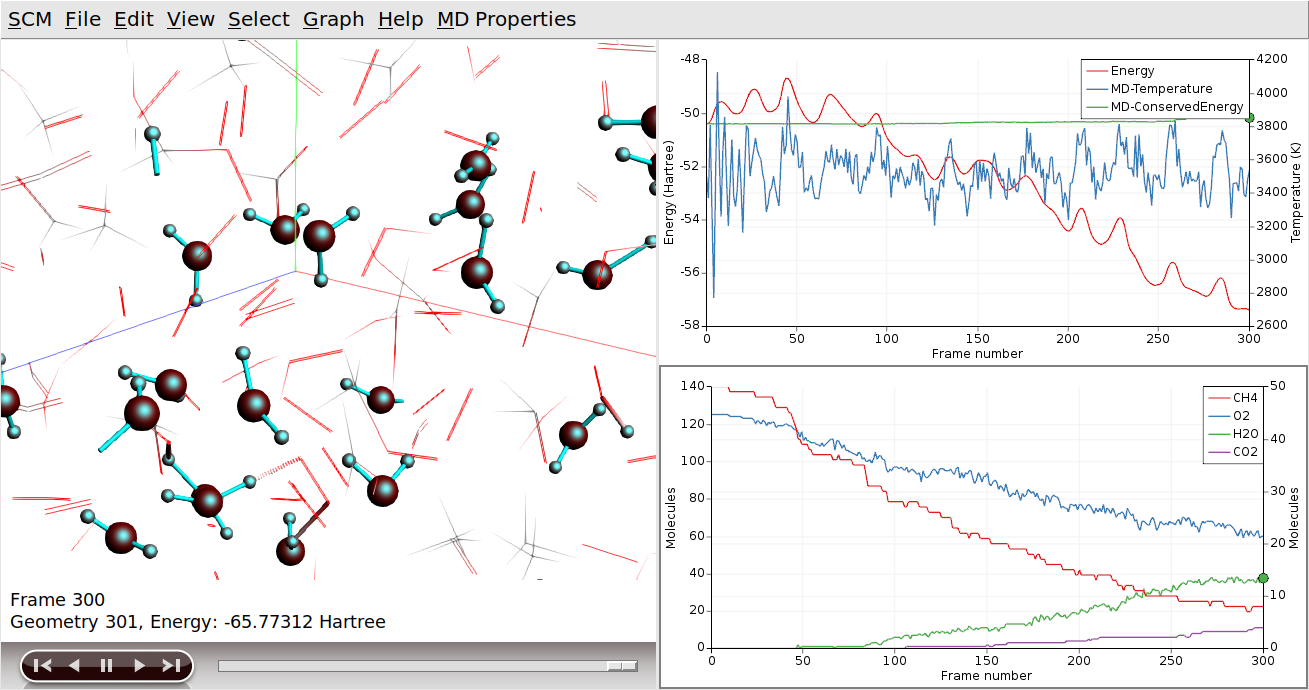
If we wanted to focus on both of our products at the same time, we can use the “Show” toggle in the Molecules dialog:
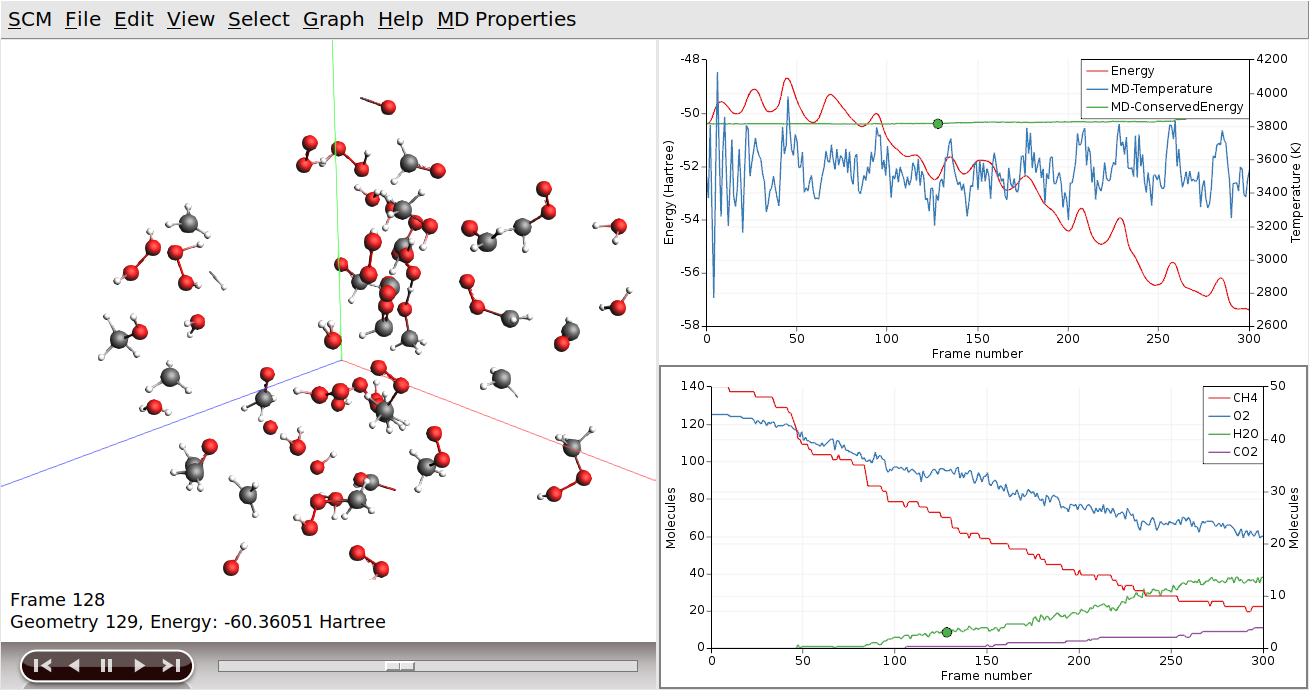
Lastly, we can also focus in on a specific region of the simulation volume: