Bond boost: Cross-linked polymers¶
In this tutorial we will show how the bond boost acceleration method can be used to drive the non-catalyzed epoxide - amine polymerization reaction with the aim of generating highly crosslinked epoxy polymer structures:
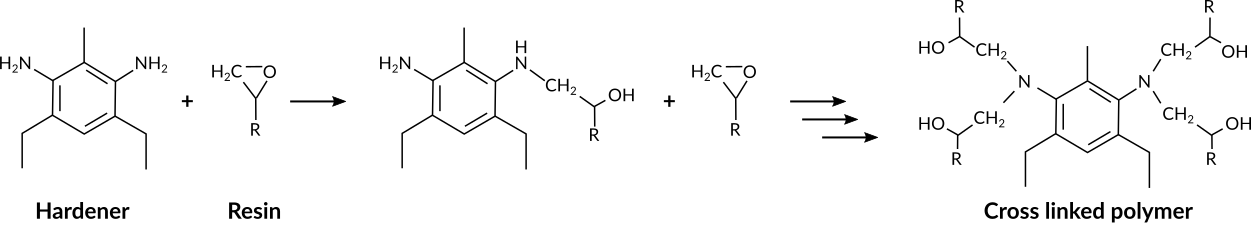
Fig. 33 Epoxide - amine polymerization reaction¶
The goal is not to capture or simulate the kinetics of the polymerization reaction, but rather to generate realistic atomistic models of epoxy polymers which can used in further simulations (e.g. for prediction of mechanical properties)
This advanced ReaxFF tutorial is loosely based on the following publications:
More details and information on other acceleration methods can be found in the ReaxFF manual:
Tip
See also the tutorial Young’s modulus, yield point, Poisson’s ratio
Setting up¶
The cross linking polymerization reaction occurs between the epoxy and the amine groups. The reaction is depicted in its simplest, non-catalyzed form in Fig. 33.
Throughout this tutorial the epoxy resin will be BisF whilst DETDA will be used as the hardener (amine):
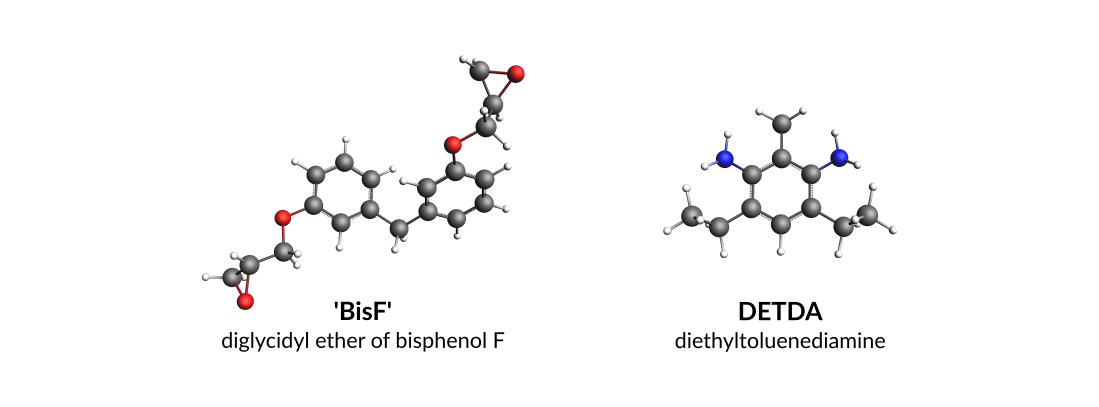
You can either draw the 3D structures of these molecules with the GUI or download the xyz files:
Use the builder functionality in AMSinput to fill a periodic box with one DETDA and two BisF molecules:
 →
→  .
.20, 20 , 20 angstrom2 and 1 for BisF and DETDA respectively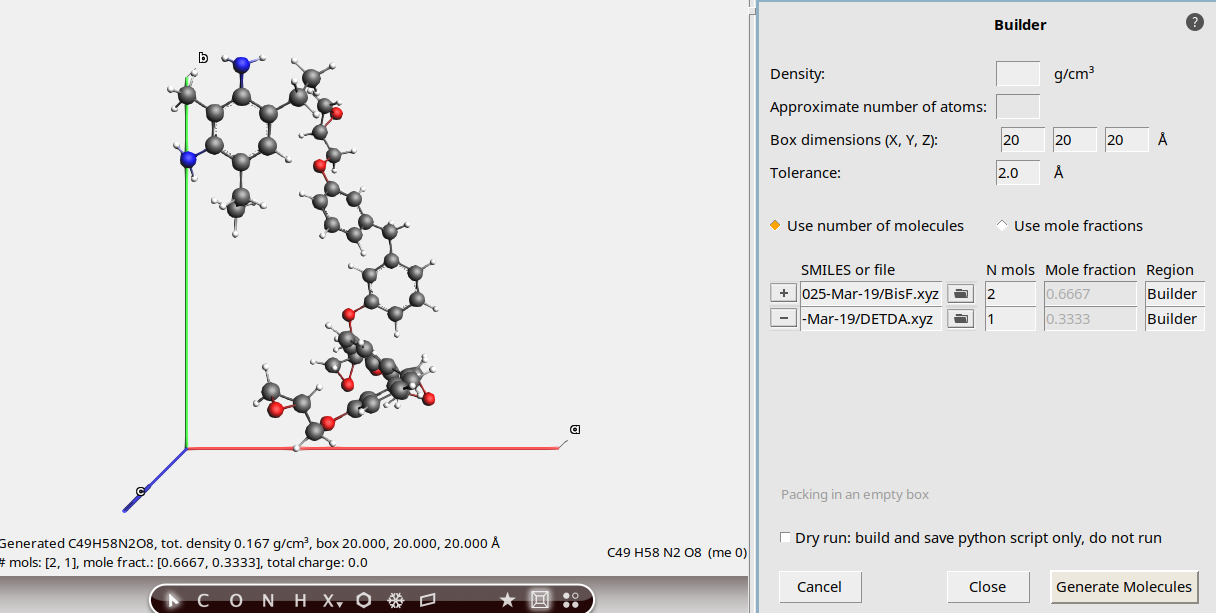
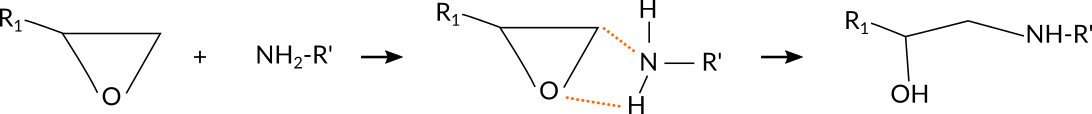
To accelerate a reaction with the bond boost method, ReaxFF needs to be provided with a set of atom distances that define a preliminary complex which could lead up to the transition state of the reaction. If this complex is formed during the dynamics, external forces are applied to support bond breaking and bond formation for a user defined set of bonds. This ‘boost’ lasts for user defined time during which the reaction may or may not occur.
For the current reaction, a simple, yet effective set of atom distances are those between the N atom and the terminal C-atom of the epoxy group as well as the distance between the O-atom of the epoxy group to whichever H-atom of the amine group is closest:
It is possible to distinguish atoms depending on their chemical environment, e.g. the terminal C-atom of the epoxy group, but the information needs to be provided via the regions model of ReaxFF by the user. To distinguish the terminal C-atom from all other C-atoms in the system, a CT region needs to be setup:
CTCT region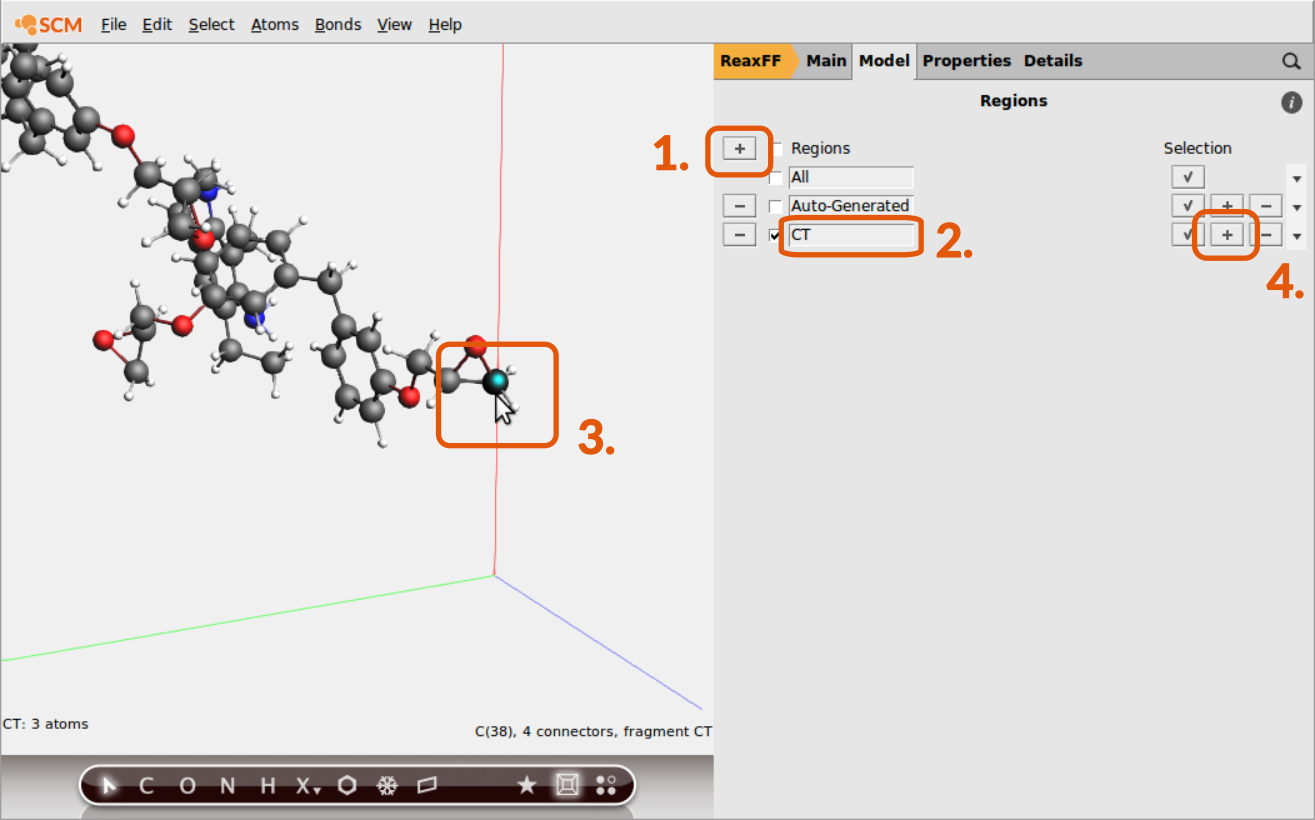
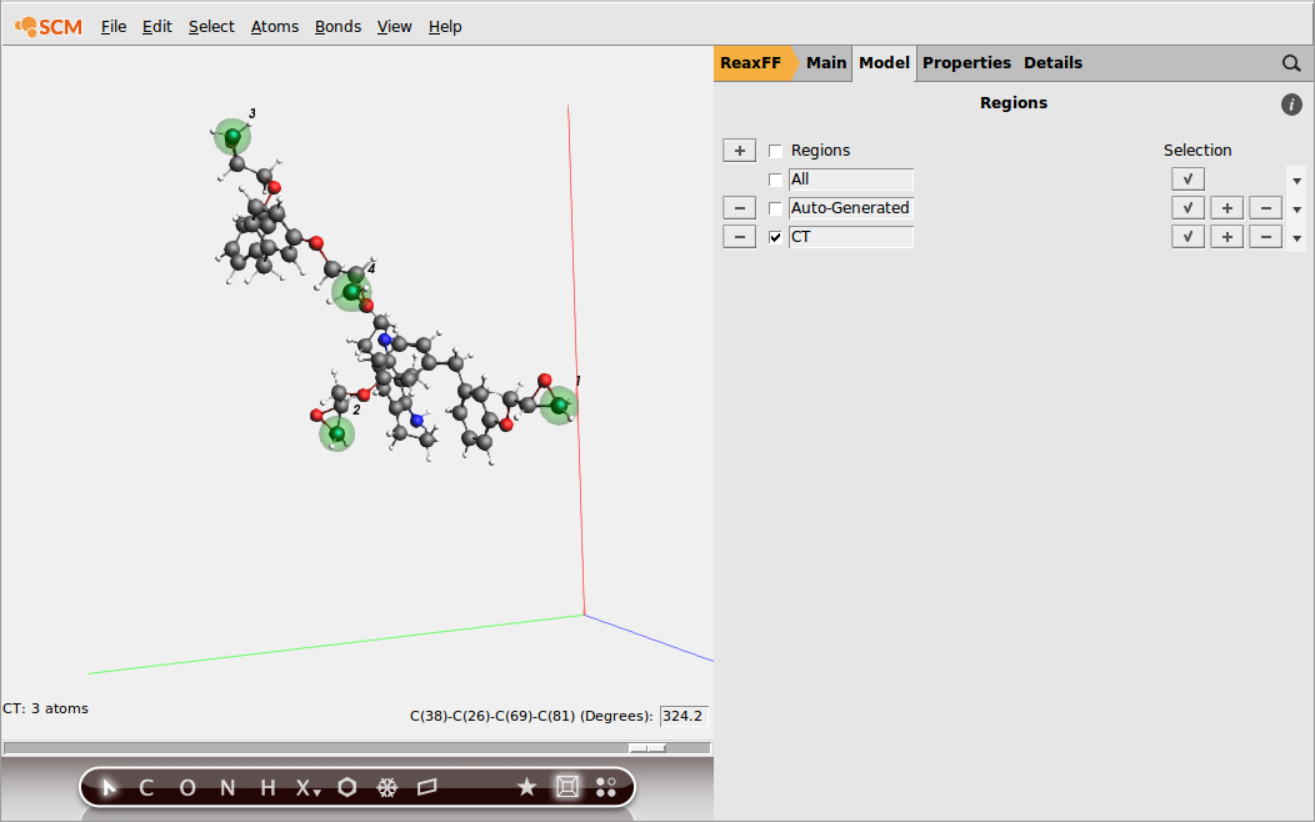
Repeat the last 2 steps to add all other terminal C-atoms (4 C atoms in total) to the new CT region
Tip
You can hold down the SHIFT key to select multiple atoms in the molecule view.
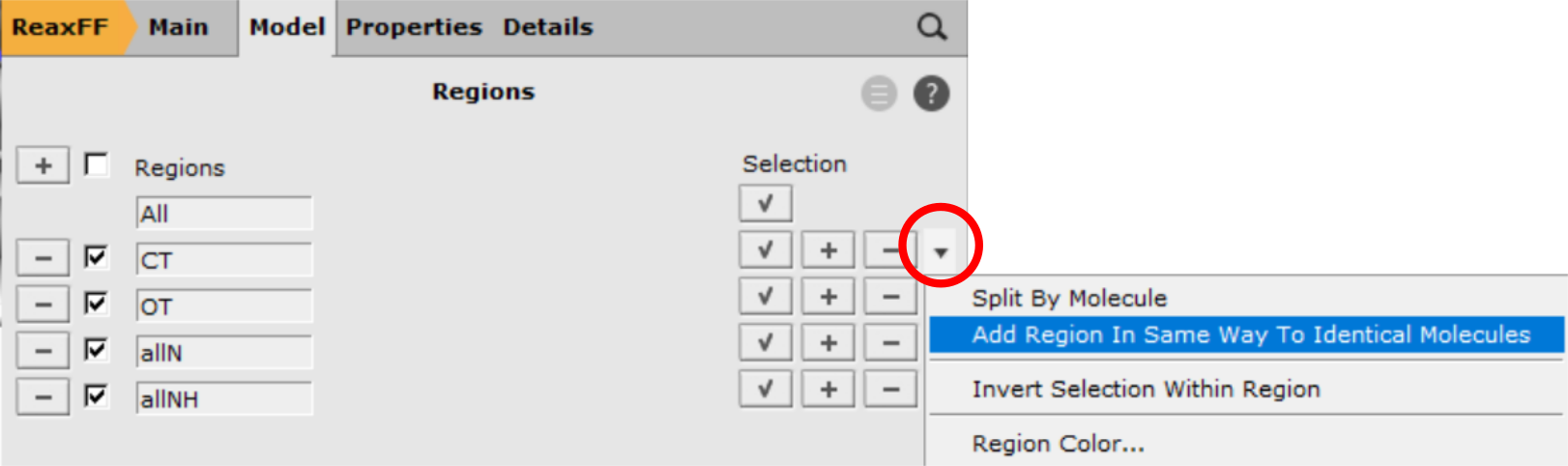
Tip
If you have multiple identical molecules, the region can be copied over in the Regions panel using the ‘Add Region In Same Way To Identical Molecules’ option from the dropdown menu on the right.
Now the subset of terminal C-atoms can be distinguished via referring to the region CT as we’ll see later. The other regions are consisting of all N-,O- and H-atoms. To set up a region with all N-atoms:
allNallN region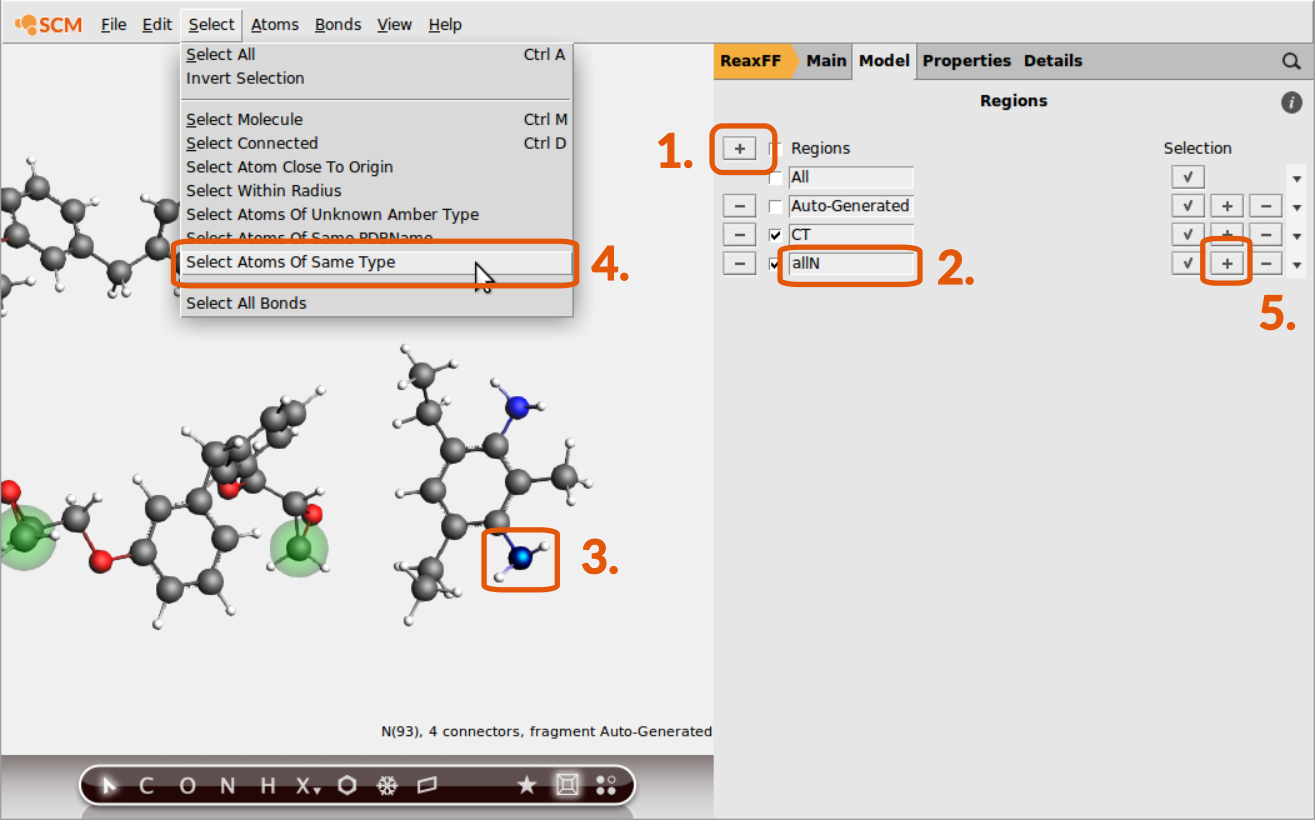
Tip
Make sure that the atoms of the previous region assignment are not selected anymore when assigning a new region. Trying to assign one atom to multiple regions is a common source of error.
Continue setting up regions containing all O- and H-atoms respectively:
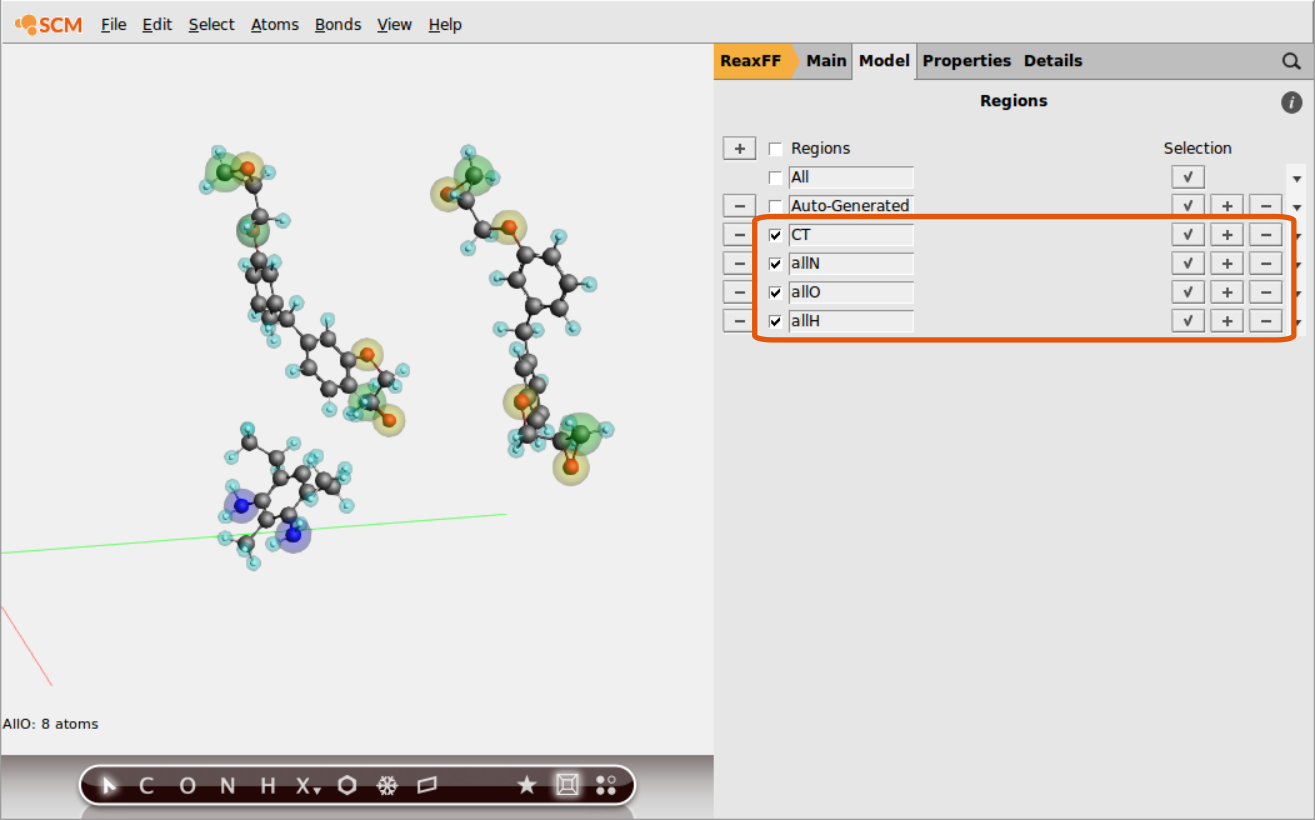
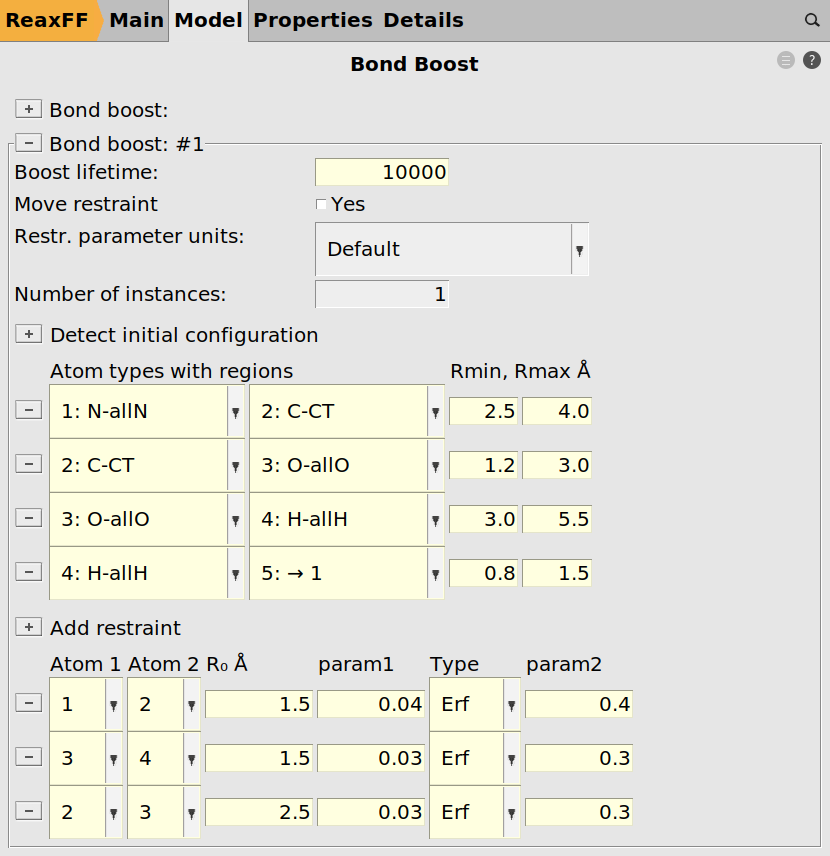
Once the regions are set up, we can use them to define a tracking regime for the bond boost. The set of atom distances that will switch on the boost once all criteria are met is
to set this up in the GUI
10000 for the Boost lifetime2.5 and Rmax to 4.0Continue with assigning the other tracking options:
C-CT / O-allO
1.2-3.0O-allO / H-allH
3.0-5.5H-allH / →1
0.8-1.5
Note
Note the last atom type is set to be →1 to specify that it must be the initially detected Nitrogen atom, i.e. the one close to CT.
The boost settings are chosen such that the following logic is implemented
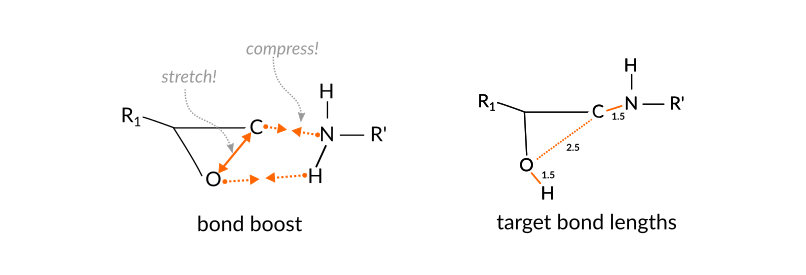
the aim is to support the breaking and forming of bonds but still allow the reaction to fail.
Note
The above settings have been chosen empirically. A more thorough assessment of energetics based on ab initio calculations is presented in the publication by A. Vashisth et al.
The bond boost options are defined in the lower part of the bond boost panel
1.50.04, the type to Erf and param2 to 0.4.Note
For the Erf restraint type, the first parameter (param1) is the force constant k, while the second parameter (param2) is the maximum allowed force F∞. The maximum allowed Force, F∞, is the most important parameter. It is important that it’s not too large because it may just rip the reactants apart if the force is too large. We found, for example, that with 0.5 it’s often ripping them apart, while with 0.3 it’s much less likely but still possible. With 0.2 the reaction takes longer but should give more reasonable results in the end. The force constant value seems to be much less essential.
Continue with assigning two more boosts:
3 / 4
1.5,0.03,0.302 / 3
2.5,0.03,0.30
Execution and visualization¶
After completing the setup of the bond boost in the previous chapter, we can test the boost by running a small NVT trajectory. Choose the following general ReaxFF settings from the main panel
dispersion/CHONSSi-lg.ff
Note
This tutorial employs the dispersion corrected force field, that has not been optimized for usage with the bond boost.
This force field has been used successfully in the study by M.S. Radue et al.
The results might be improved by using the force field CHON2017_weak_bb.ff fitted for usage with the bond boost method (A. Vashisth et al. ).
However, note that switching the force field will require tweaking the bond boost settings.
Open the MD settings:
 next to Molecular Dynamics
next to Molecular Dynamics40000
Open the thermostat settings:
next to Thermostat next to Thermostat
next to Thermostat100.0 fs500.0 K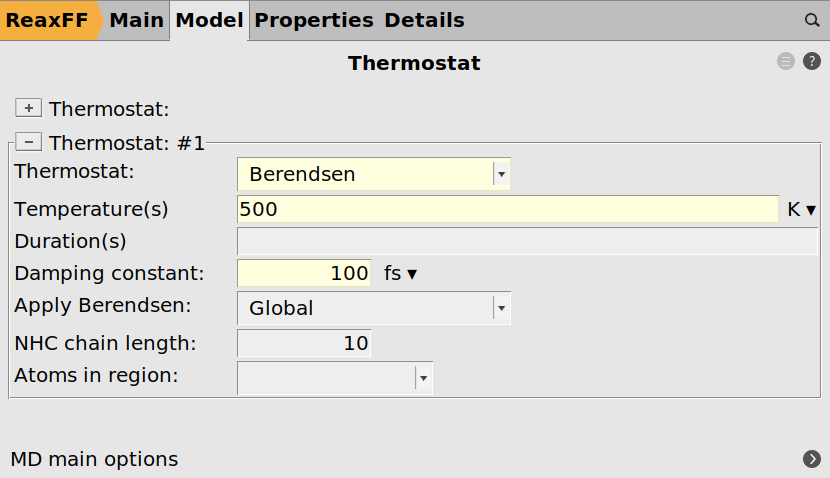
Save and run the calculation:
Open AMSmovie and follow the calculation of the trajectory, you should see at least one successful cross linking reaction. The boost periods are visible as kinks in the energy graph
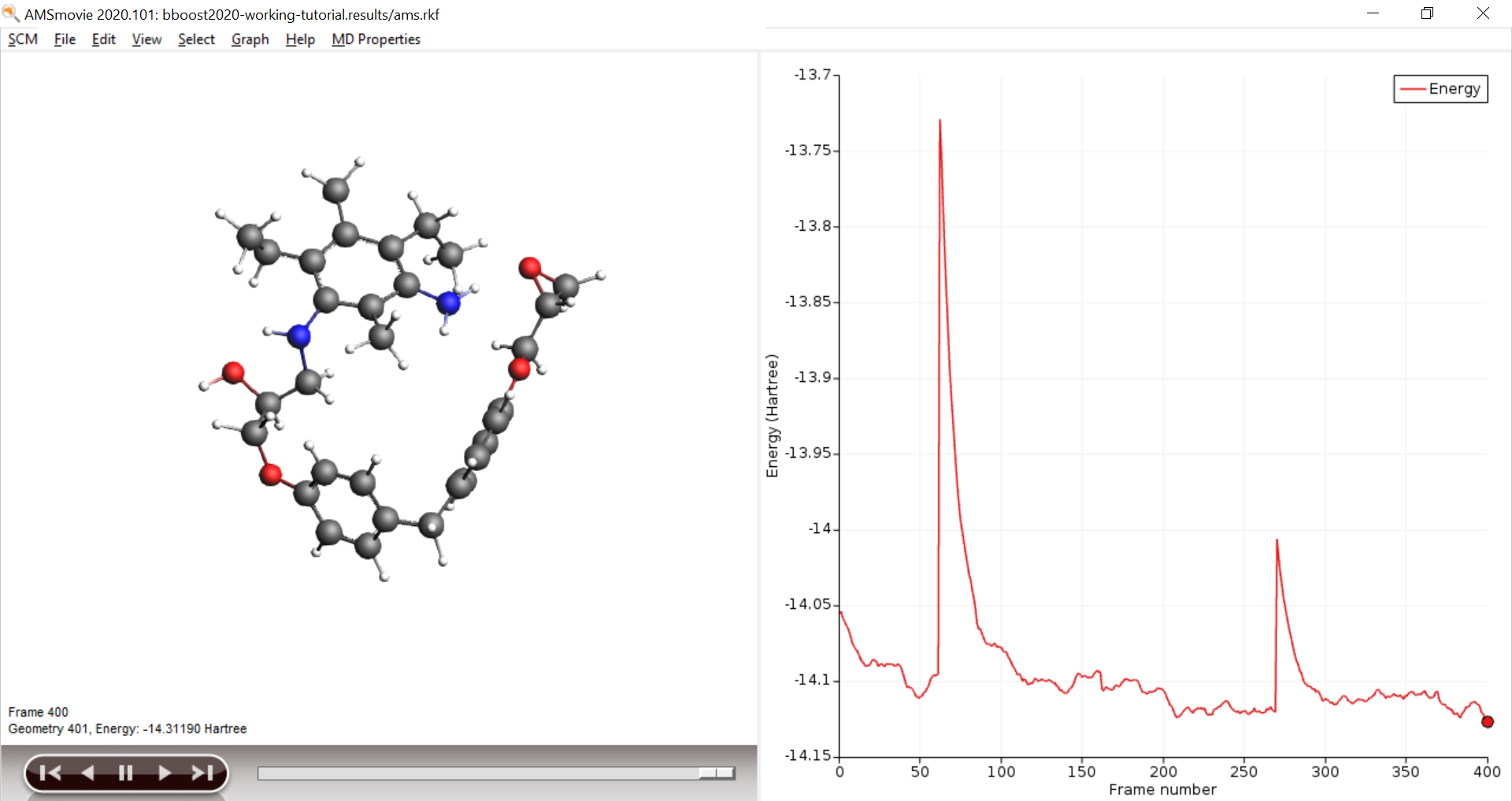
Scaling it up: Generate large Polymer structures¶
For simulation of polymer properties much larger structures are needed. In the following chapter, we will explain how to scale up the bond boost simulation to more realistic systems up to several thousand atoms.
Open a new AMSinput window
Open the Packmol builder to create a larger initial structure. Set up a periodic box with with 20 BisF epoxy and 10 DETDA hardener molecules.
→ .32, 32 , 32 angstrom button and open the file DETDA.xyz20 and 10 for BisF and DETDA respectively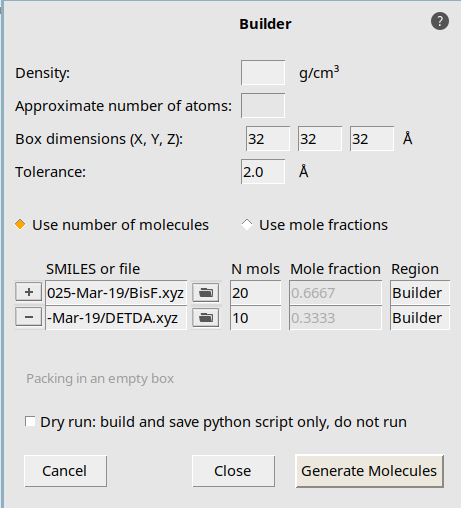
Next we need to assign regions to one BisF and one DETDA molecule. One molecule of each species will suffice. Assign the following regions to one of the DETDA and one of BisF molecules:
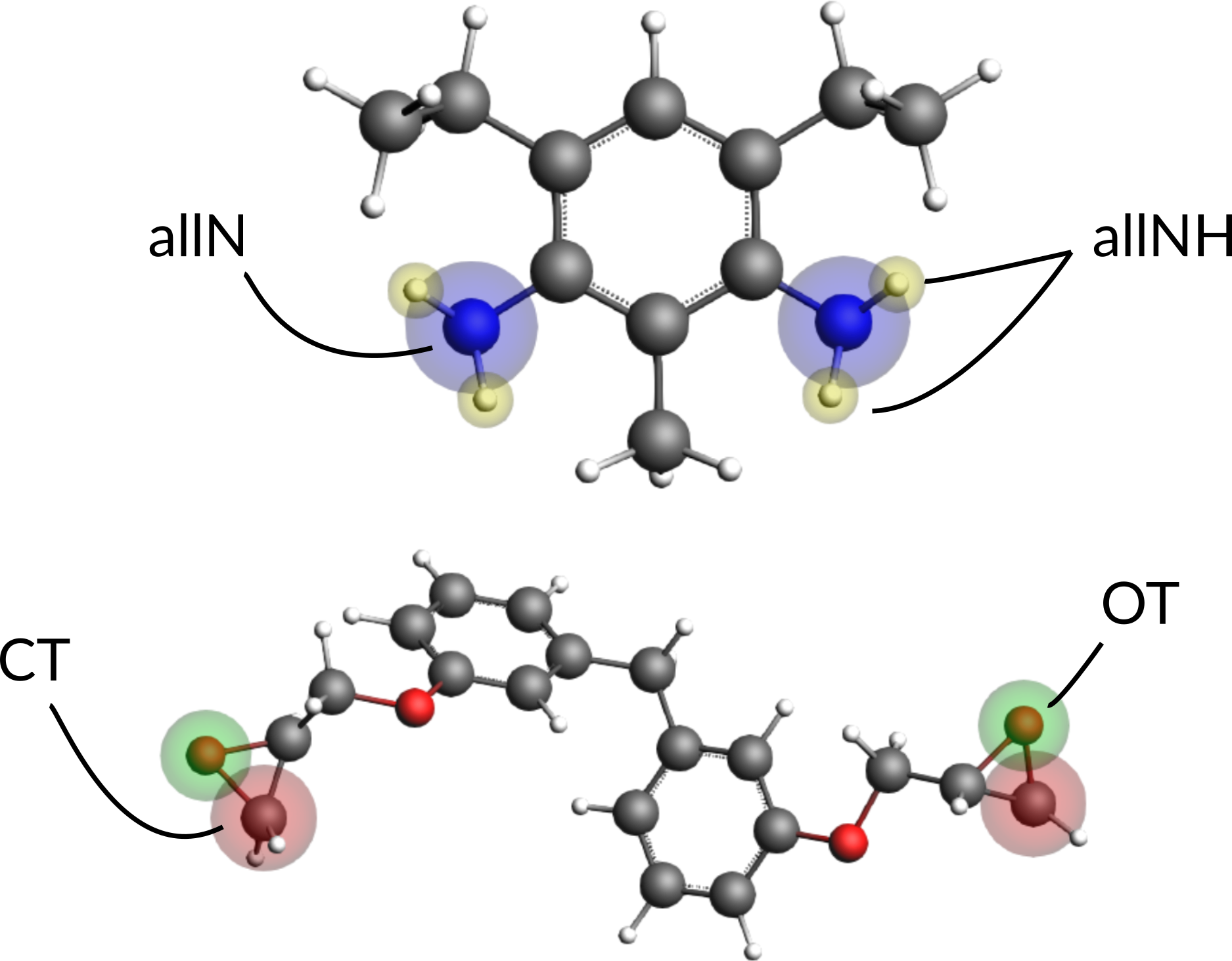
To assign the same regions to all other molecules automatically
Tip: assign regions before packing
You may also assign the regions before packing into a box. That makes it easier to pack multiple boxes with different sizes, concentrations, etc.
To do this, open BisF.xyz, assign the regions, and export the system using File → Export System → .in. This format stores the region information.
Repeat for DETDA.
In the Builder dialog, browse to the BisF.in and DETDA.in files instead of the .xyz files.
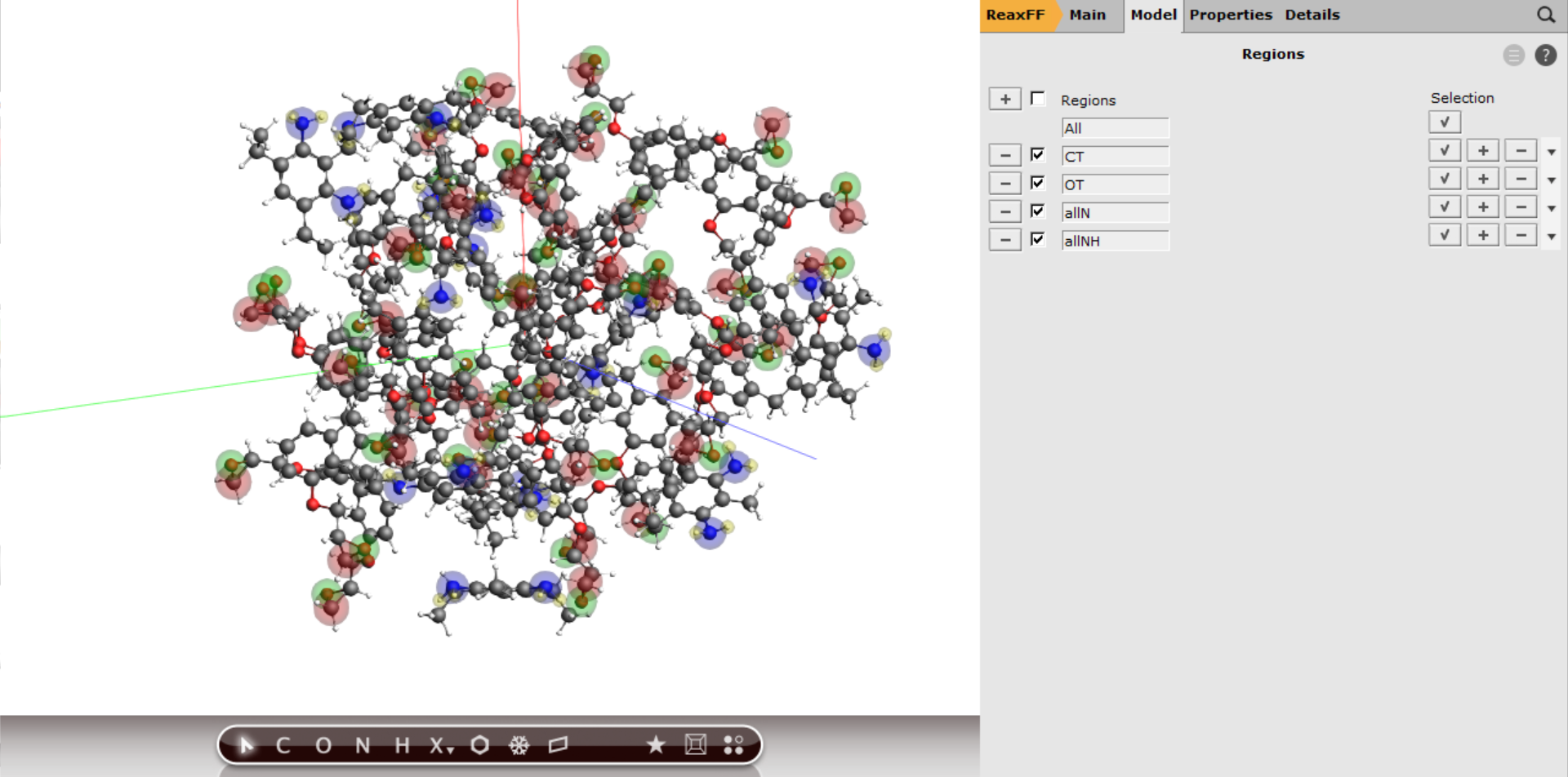
Your periodic box with all the regions should now look like this
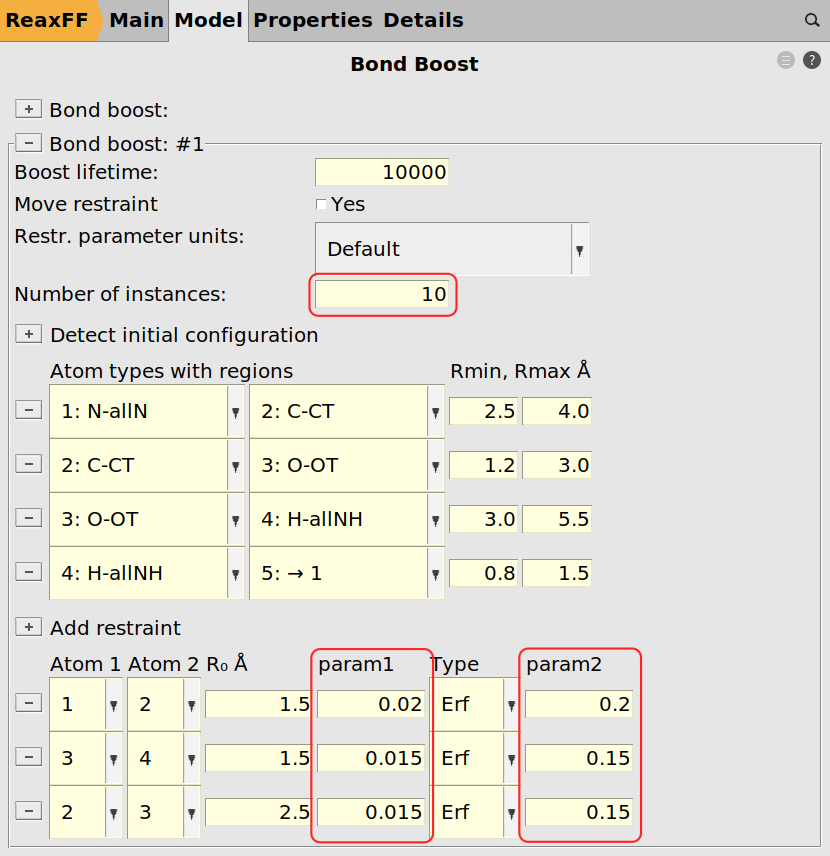
Next we can assign the tracking for the bond boost using the same logic as before with the exception of requesting more bond boost instances and softer restraints. The instances refer to the number of simultaneously boosted reactions. Since we have ten amines, i.e. ten reaction sites, we could in principle expect ten simultaneous reactions (not counting double reactions on the same amine group). The restraints are softened to prevent the molecules from tearing apart when multiple boosts are active at the same time.
Now only the MD settings need to be set.
dispersion/CHONSSi-lg.ff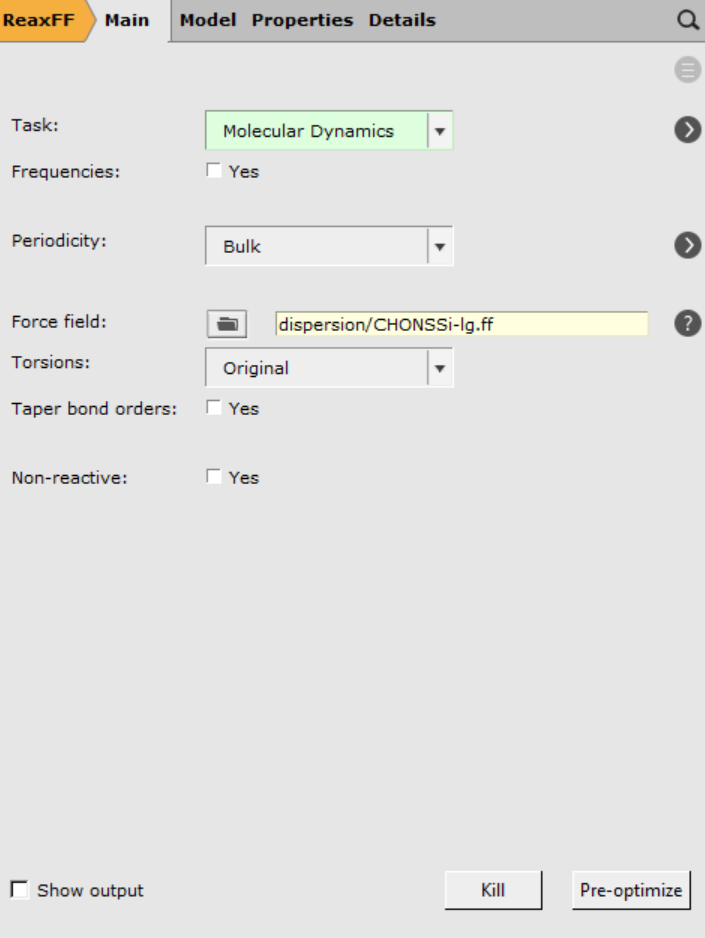
This time we shall run the simulation for a longer time. To increase the efficiency of the calculation, we will lower the framerate from writing a snapshot every 100th to every 1000th MD step.
Open the MD settings:
next to Molecular Dynamics2500001000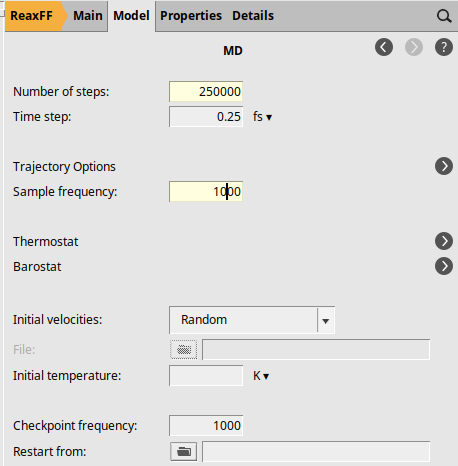
We use the same thermostat as before but will add a barostat:
next to Thermostat next to Thermostat100.0 fs500.0 K
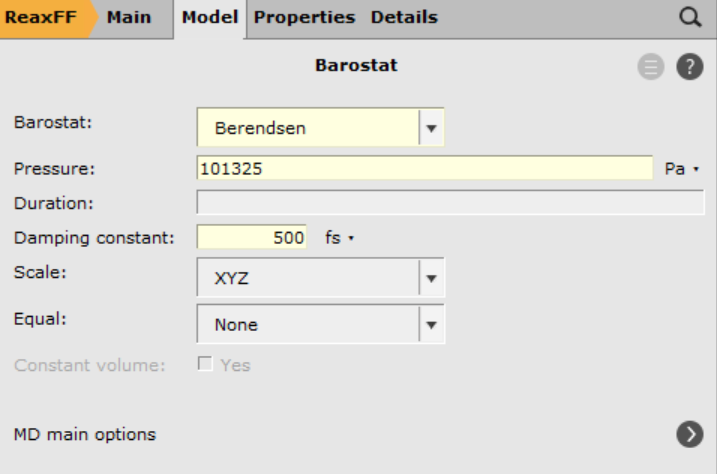
next to MD main options next to Barostat101325 Pa (1 atm)500.0 fsSave and run the calculation (may take several hours):
Analysis: Calculate the density and cross-linking ratio¶
The trajectory can be monitored in AMSmovie, though it becomes increasingly tough to spot the reactions with such huge systems. For this reason, we provide a special analysis tool that allows you to monitor the progress of the systems cross-linking via the cross-linking ratio.
cross-link-density.pyNote
You can find more information on PLAMS and AMS scripting in the Getting started section of the scripting documentation.
To run the script execute the following command in the command line (replace myjob.results with the path to your results directory):
$AMSBIN/amspython cross-link-density.py myjob.results
The script will print the results onto the command line something like:
...
( 241/ 251) 48 0.700
( 242/ 251) 48 0.700
( 243/ 251) 48 0.700
( 244/ 251) 48 0.700
( 245/ 251) 48 0.700
( 246/ 251) 48 0.700
( 247/ 251) 48 0.700
( 248/ 251) 48 0.700
( 249/ 251) 48 0.700
( 250/ 251) 48 0.700
Final density: 163.418 kg/m^3
Cross linking ratio: 0.7
The values in parentheses refer to the current analyzed frame and the total number of frames.
The second column is the total number of C-N bonds found in the system at that frame
The third column is the cross-linking ratio computed as the ratio between newly formed CN-bonds and number of theoretically possible CN bonds (two new bonds per amine group).
The MD simulation depends on random numbers, therefore your results may differ from the ones discussed here.
Note
Obviously the density has not yet converged to the experimental value. Whether it’s possible to just extend the simulated timescale, depends on the system. Often it helps to run one or several simulated annealing runs with such a cross-linked polymer structure to eliminate local density hot spots or vacuum bubbles in the structures. An enlightening discussion can be found in computational details of A. Vashisth, C. Ashraf, W. Zhang, C. E. Bakis, and Adri C. T. van Duin, J. Phys. Chem. A 2018, 122, 32, 6633-6642 (2018)
You may also run the simulation at a much higher pressure, for example, 10 kbar.