Commemorating Tom Ziegler’s contributions to Density Functional Theory
The news of Tom's sudden and peaceful passing in March 2015 came as a shock to his close friends and collaborators. Here we commemorate Tom with a short and incomplete overview of Tom's contributions during his lifelong dedication to theoretical chemistry, and DFT in particular.

The news of Tom’s sudden and peaceful passing in March 2015 came as a shock to his close friends and collaborators, particularly in Canada and Amsterdam (SCM, theoretical chemistry).

Tom Ziegler, 1945-2015
With Tom, the entire computational chemistry community loses a unique personality and scientist. He possessed a special combination of qualities: thorough theoretical insight, chemical intuition, endurance in working out equations, coding them and finally applying to real-life chemical problems. As witnessed by his entry on the International Academy of Quantum Molecular Science (IAQMS) website, Tom more than succeeded in this, receiving a large number of awards (the WATOC Schrödinger medal being one of many) and tackling problems in industrial catalysis in collaborations with various companies.
His achievements have been highlighted in two(!) special issues of the Canadian Journal of Chemistry, including a tribute and career overview by Arvi Rauk and a personal account of two former group members Heiko Jacobsen and Tom Woo about the early days of DFT in chemistry when Tom already referred to DFT as “Das Future Tool”.
Here we commemorate Tom with a short and incomplete overview of Tom’s contributions during his lifelong dedication to theoretical chemistry, and DFT in particular.
Establishing DFT as a mainstream tool in chemistry
Tom was one of the pioneers in applying DFT to chemistry, while the mainstream chemical community did not want to have anything to do with Density Functional Theory, an odd non-ab initio theory physicists used to describe the electronic structure of solids.
His visionary Chem. Rev. paper of 1991, Approximate density functional theory as a practical tool in molecular energetics and dynamics, is cited over 2000 times. In that same year, Density functional theory as a practical tool for the study of elementary reaction steps in organometallic chemistry, is the first paper to show up in Web of Knowledge with the keywords ‘DFT and Chemistry’. It takes a while for DFT to infuse in the chemical community, but application papers by Tom and others, in particular involving transition metal chemistry, slowly convinces chemists to start using DFT and by the late nineties, DFT has firmly established itself as a mainstream tool for chemistry.
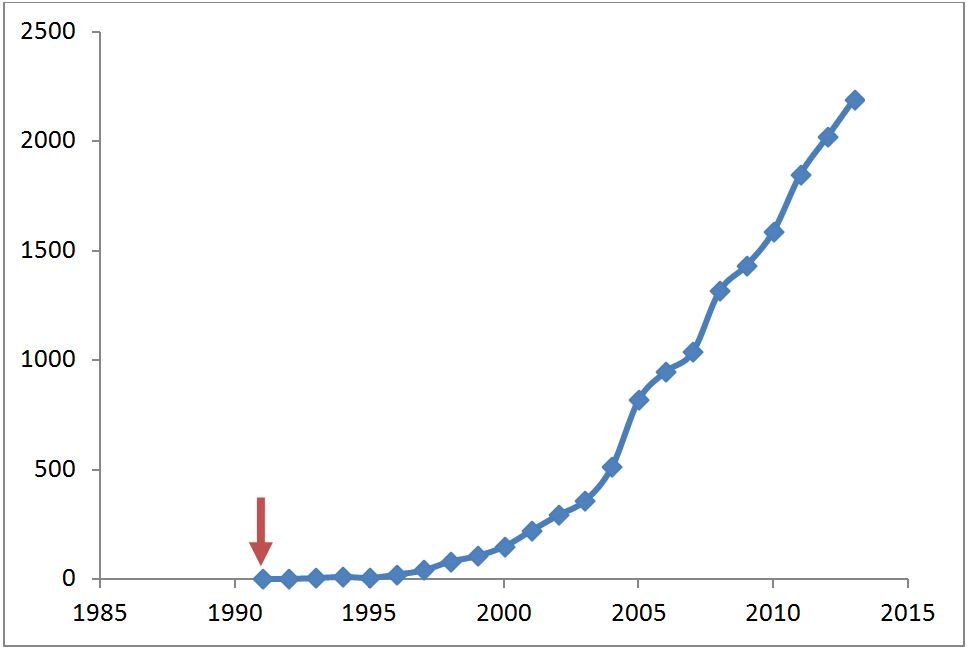
Number of papers in web of knowledge database with the topic ‘DFT and Chemistry’. No papers appear before Tom Ziegler’s 1991 seminal paper (red arrow).
Early days: DFT with atomic orbitals
In 1976 Tom started to collaborate intensively with the Amsterdam group using the then just developed DFT method (or Xa, or Hartree-Fock-Slater) which used an atomic basis set approach, density fitting and special numerical techniques. Two seminal papers appeared in 1977: Ziegler & Rauk, On the calculation of bonding energies by the Hartree Fock Slater method, Ziegler, Rauk, Baerends: On the calculation of multiplet energies by the Hartree-Fock-Slater method. The first paper was both technically and conceptually very important. It offered a way to calculate bond energies even with the, at that time still limited, numerical accuracy of the calculations, and at the same time introduced a symmetry based decomposition of the bond energy, the extended transition state (ETS) scheme. The second paper, often cited as the origin of the ∆SCF method, resulted from the interest in spectroscopic properties which after three decades led to Tom’s original approach to excitation energy calculations (CV-DFT). During his postdoc time in Amsterdam he applied the just developed relativistic method of ADF to chemical bonding of heavy element compounds, resulting in very fundamental new insight in the physical origin of the relativistic bond contraction (quipped “ the Dutch revolution” by Pekka Pyykkö), (Relativistic effects on bonding, 1981; Calculation of bond energies in compounds of heavy elements by a quasi-relativistic approach, 1989).
Continuing developments: spectroscopy, relativity
Tom continued to develop and apply novel DFT techniques. Most notable are his contributions to magnetic resonance spectroscopy (NMR and ESR) (e.g. Calculation of NMR Shielding Tensors Using Gauge-Including Atomic Orbitals and Modern Density Functional Theory, 1995; Calculation of the G-tensor of electron paramagnetic resonance spectroscopy using gauge-including atomic orbitals and density functional theory 1997). The unique emphasis on relativistic effects and the ensuing applicability to heavy element compounds made these relativistic approaches to spectroscopic properties such as NMR, EPR, but also UV/VIS (e.g. spin-orbit coupled TDDFT with Fan Wang, later applied to OLEDs), practical and applicable to large and heavy systems.
Other theoretical developments: QM/MM, COSMO(-RS), BAND, ETS-NOCV, CV-DFT
Tom’s broad theoretical grasp and scientific interest also transpires in his implementation of QM/MM (Implementation of the IMOMM methodology for performing combined QM/MM molecular dynamics simulations and frequency calculations, 1998), the conductor-like screening model (An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package, 1999) and the real solvents (COSMO-RS) thermodynamics model based upon that (An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Part II. COSMO for real solvents, 2009) and various contributions to the periodic DFT code BAND (gradients, NMR, EPR).
The natural orbitals for chemical valence refinement (ETS-NOCV) of Ziegler and Rauk’s extended transition state theory, in collaboration with Mitoraj and Michalak (A Combined Charge and Energy Decomposition Scheme for Bond Analysis, 2009), is Tom’s latest contribution to gaining in-depth insight in chemical bonding.
Aside from a large number of other important contributions to the computational chemistry field, Tom spent his last years on implementing and perfecting constricted variational theory (CV-DFT). This promising method for excited states overcomes some of the notorious problems in TDDFT (The implementation of a self-consistent constricted variational density functional theory for the description of excited states, 2012). He strived to bring this last big project to fruition as a mature, robust and practical DFT approach to excited states. Although unfortunately Tom will not be there to witness it himself, his inspiration to coworkers and the community on a whole will surely see to the realization of this latest vision.
Catalysis
Tom has applied the computational chemistry toolbox, to which he contributed so extensively, to many problems in chemical bonding (notably in transition metal and organometallic chemistry) and reactivity. He published massively in the field of catalysis, which he took as the “core” of chemistry, organizing his development work around such goals as detection of species by spectroscopy and elucidation of reaction mechanisms by reliable computation of reaction barriers and reaction paths.
Some of the papers by Tom with more than 500 citations can be found on Google scholar with the Chemical Reviews paper cited over 2,000 times. This put Tom among the 100 most-cited chemists worldwide and second best cited chemist in Canada. According to the statistics on Tom’s website, he published close to 500 papers, being cited more than 25,000 times leading to an H-index of 72.
For the ADF package, Toms influence can hardly be overestimated. In the ADF manual alone, his name occurs 64 times in the References section, more than one out of seven of the total.
His impact will continue through his publications, his software, but also his former PhD students and post-docs, several of whom went on to establish their own thriving research groups. Others have moved on to research positions in industry or other careers.
Amsterdam, 31 March 2015
Evert Jan Baerends
Stan van Gisbergen














