Embedded MgO cluster
In this tutorial you will learn how to create a cluster model from the bulk and how to define custom elements (J-elements) which can have any nuclear charge and basis set.
Choose a ‘New Input’ and switch to BAND from ADF in the drop-down from the yellow menu bar. Now you will build a periclase (MgO) crystal. To start building a crystal: click on the structure tool icon (ice crystal), or alternatively click Edit → Crystal . Select NaCl from the Cubic Crytals. Change the lattice constant to 4.2, Na to Mg, and Cl to O and click OK.
Next we will cut a (001) surface slab from the bulk. Click the slicer button (the parallelogram next to the ice crystal) or choose generate slab from the Edit → Crystal menu bar. Make sure you select ‘Cartesian’ next to the Miller indices and cut a 4-layer (001) surface slab.
We’ll create a 2√2 x 2√2 supercell from this slab: Edit → Crystal → Generate Super Cell and set the matrix to 2, -2, 2, 2. (Alternatively you can first create a √2 x √2 supercell from the presets, followed by a 2 x 2 super cell). Turn off the periodic view and lattice vectors in the View → periodic menu, and Remove Bonds in the Bonds menu.
Before making this a cluster, we first map all atoms within the unit (super) cell: Edit → Crystal → Map Atoms To Unit Cell… This should now look like a neatly arranged 4 x 4 x 4 cluster:
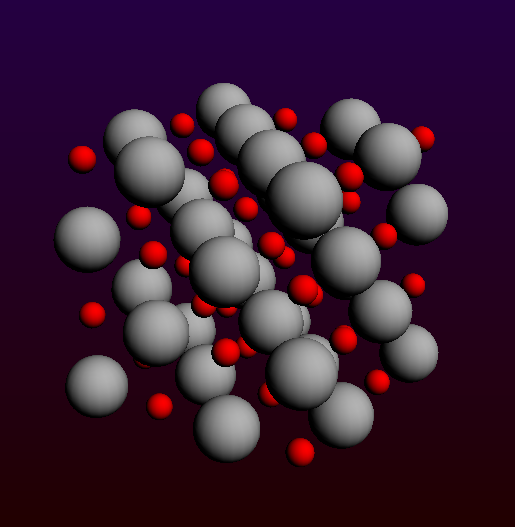
4x4x4 MgO (001) slab to be turned into a cluster
Switch to ADF input (click on the yellow menu bar ‘BAND’ and select ADF from the drop-down): We have turned our 2D-periodic slab into a Mg32O32 cluster. For a cluster model to simulate a surface the edges need to be terminated so as to best recover the properties of the surrounding solid. In ADF one can utilize (a combination of) various options:
- point charges
- (polarizable) force field
- capping atoms (IMOMM or AddRemove)
- custom elements (J-elements)
Here we give an example of how one might employ custom elements in ADF. In ADF’s fragment approach, one can assign whatever charge or basis set to an element. To model an ionic system like MgO, one would typically aim to reduce the charges at the cluster edges to counteract the undercoordination of these atoms and thereby avoid an artificial build-up of charges at the edge. Probably the expert user would most quickly set such a calculation up via a set of scripting tools, but in principle it is also possbile in the GUI as demonstrated below.
We’ll take a (MgO)4 cube as our full-electron cluster and customize the remaining 56 atoms at the edges of this surface cluster. In he bulk, every atom is hexacoordinated and we will attempt to change the elements as much as possible in accordance with that.
We have 3-, 4-, and 5-coordinated species at our edges. For each missing coordination we will remove 1/6th of the full ionic charge, i.e. 1/3rd electron. The charge of elements selected can be adapted in the ‘Atoms details’ pane, which you will find under Atoms → Details (Color, Radius, Mass …). In the Atom Details pane tick the Znuc box, a specialist option, so that we can assign charges in the ‘Q’ column. Give 3/4/5-coordinated Mg atoms a charge of 11/11.3333/11.66667 and 3/4/5-coordinated O atoms a charge of 9/8.6667/8.3333.
You can also color the appointed charges by Properties → Color atoms by → Znuc . The custom elements will be given a large frozen core basis, which we can set in the Main panel by clicking on the … button next to the basis set drop-down menu. We’ll choose an all-electron basis set for our (MgO)4 cube by selecting AE → TZ2P from the drop-down menu next to Mg and O.
The GUI has automatically assigned names to your J-elements, corresponding to the element they originated from and their first occurence in the structure. For the J-elements elect a 2p core and DZ basis for Mg and a 1s core with DZP basis for O:
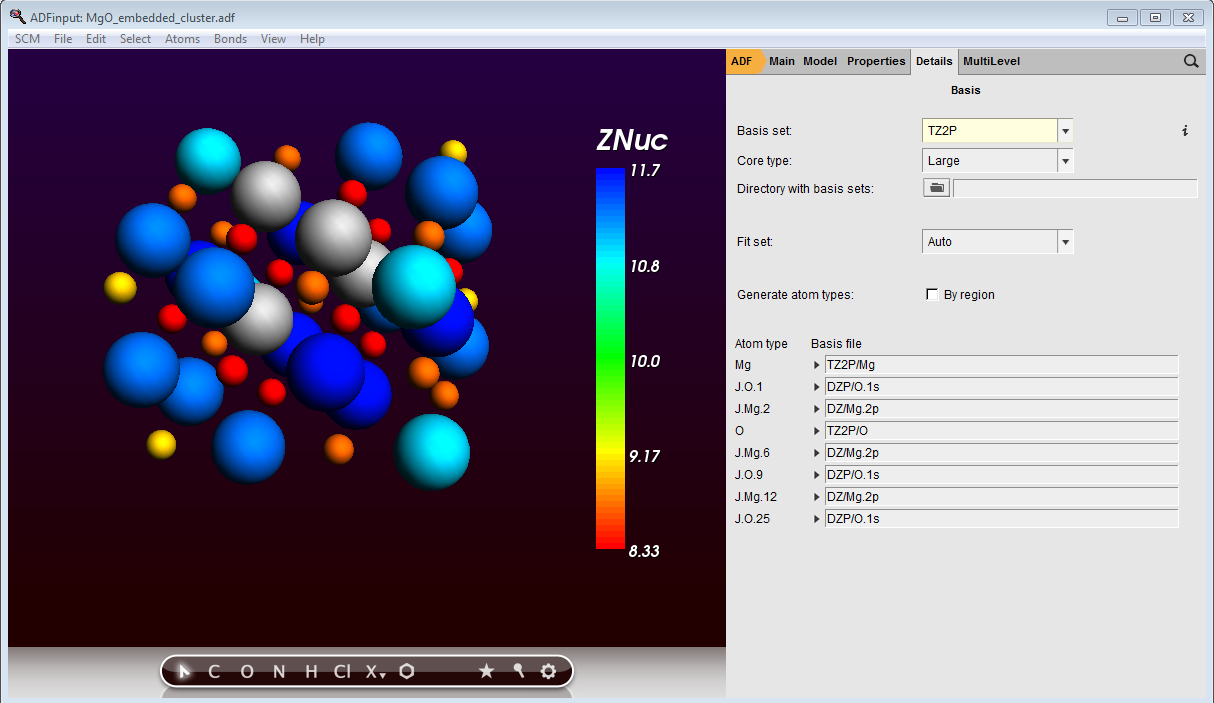
MgO cluster embedded in custom elements with fractional charges and custom basis sets
If we want to study the adsorption of, for instance, water on the MgO surface, it is probably best to select a dispersion-correct functional like BP-D3(BJ) in the Main panel from the drop-down menu next to ‘XC potential in SCF:’
An integration accuracy higher than the default would be advisable for such systems. After having made these selections, press Ctrl/Cmd + R to start your calculation (after you have been prompted to save your input).
You can also use the run file that results from following these tutorial steps and further adapt it as you wish.
After your calculation has finished, open up ADFView and have a look at the different charge types (Properties → Atom info. In ADFView the custom elements are displayed with much smaller radii, making it easier to focus on the ‘all-electron’ part.
Also check in ADFView (under Properties) that the HOMO of our embedded cluster is located at the surface of our all-electron (MgO)4 cube. It is an easy excerise to show that for a 4 x 4 x 4 MgO cluster without these custom atoms at the cluster edges, the HOMO is located at the cluster edges. Therefore an unterminated cluster is unsuitable to study the chemistry of a (001) surface:
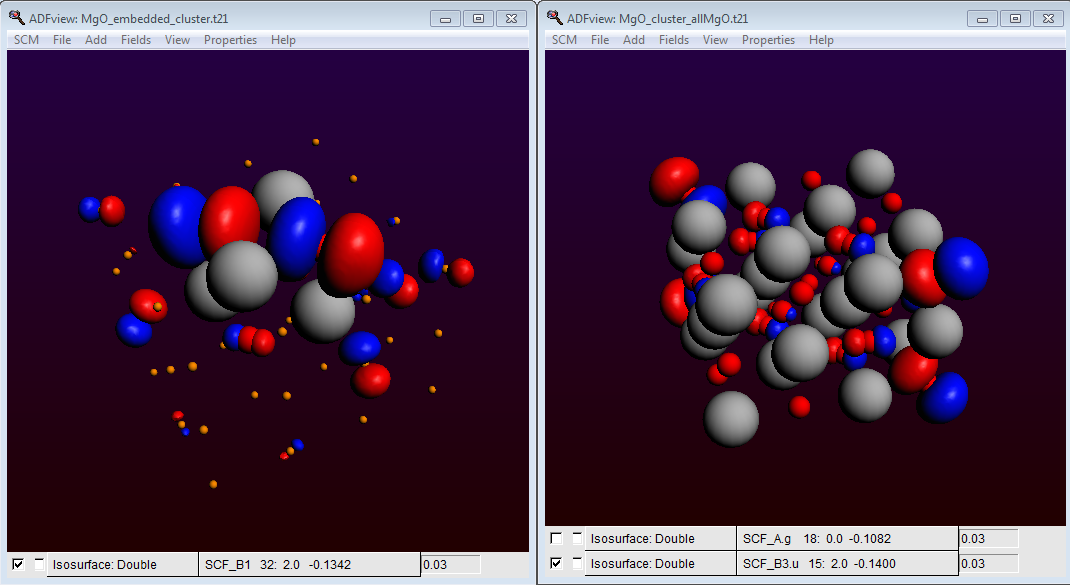
HOMO of MgO: orbital localization in the embedded cluster (left)
To model your own surface cluster with custom elements, have a look at the documentation. With scripting tools to determine the local coordination number one could quickly assign J-element types to atoms at the edge of a cluster cut from a surface slab. In the create runs one can design entirely customary atoms. The cluster set-up can be compared with BAND calculations on the periodic 2D-structure, for instance to recover as best as possible the density-of-states in the model cluster, or the adsorption energy of a molecule.
Publications using embedding with fractionally charged atoms
Example publications using fractionally charged atoms with fractional occupations to terminate ionic clusters with pseudo-hydrogen atoms:
Adsorption of chemical warfare analogues on γ-Al2O3 surfaces, TiO2 surfaces