Example: Benzenedithiol junction: Wide-Band-Limit¶
In the wide-band limit (WBL) the coupling to the leads is assumed to be independent of energy. Therefore one does not need to calculate any self-energies. This also means that the eigenspace of the Green’s function is independent of energy. It can therefore be diagonalized in advance, greatly speeding up the calculation of the DOS and the transmission.
In the example $AMSHOME/examples/adf/green_Al/green_WBL/green_WBL.run of green, the transmission of benzenedithiol junction in the wide-band limit (WBL) is calculated. In order to model the molecule-metal interface, we do need to include a few gold layers in the calculation. However, unlike before, only a single atomic layer as the principal layer is used.
Because a single atomic layer is an unnatural configuration for gold, a minor amount of smearing is necessary to make the calculation converge. The molecule is sandwiched in between the electrodes just like before (see Fig. 2 in the example for benzenedithiol). However, this time each atomic layer of gold gets its own fragment. The reason for this configuration is that if the WBL is used on the entire gold contact the result is an an unphysical coupling to the leads; even the gold atoms contacting the molecule would have a direct coupling to the environment. A much better result can be obtained by only using the WBL on the back-most atomic layer and letting the electrons propagate naturally through the rest of the contact. Because the WBL is computationally so inexpensive, we can easily calculate the DOS and transmission for 10,000 points instead of 1000.
A comparison of the resulting transmission with the calculation with self-energies is shown in the following figure:
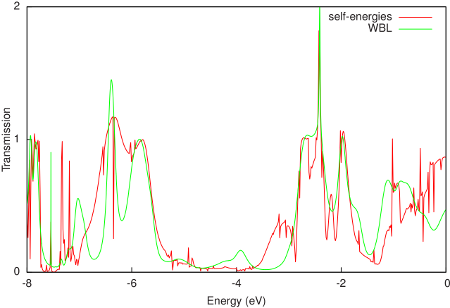
The WBL shows good agreement with the non-WBL transmission around the Fermi energy (-0.195 Hartree or -5.306 eV). Note that the quality of the WBL depends on the choice of the coupling (ETA). For this particular contact geometry we obtain good agreement for ETA = 0.02 Hartree, but a better value may be found for other electrodes. Finally, the WBL can be incrementally improved by adding more gold layers to the extended molecule. For many layers it converges to the calculation with full self-energies.
#!/bin/sh
# In the wide-band limit (WBL) the coupling to the leads is assumed to be independent of energy.
# Therefore one does not need to calculate any self-energies.
# This also means that the eigenspace of the Green's function is independent of energy.
# It can therefore be diagonalized in advance,
# greatly speeding up the calculation of the DOS and the transmission.
# In the example $AMSHOME/examples/adf/green_Al/green_WBL.run of green,
# the transmission of benzenedithiol junction, example green_BDT.run,
# in the wide-band limit (WBL) is calculated.
# In order to model the molecule-metal interface, we do need to include a few gold layers
# in the calculation.
# However, unlike in green_Au.run, only a single atomic layer as the principal layer is used.
# In green_BDT.run 3 layers of gold atoms from 1 fragment.
# In this example green_WBL.run each layer of gold atoms has its own fragment.
# Because a single atomic layer is an unnatural configuration for gold,
# a minor amount of smearing is necessary to make the calculation converge.
# The molecule is sandwiched in between the electrodes just like before
# (see the example for benzenedithiol, example green_BDT.run).
# However, this time each atomic layer of gold gets its own fragment.
# The reason for this configuration is that if the WBL is used on the
# entire gold contact the result is an an unphysical coupling to the leads;
# even the gold atoms contacting the molecule would have a direct coupling to the environment.
# A much better result can be obtained by only using the WBL on the back-most atomic layer
# and letting the electrons propagate naturally through the rest of the contact.
# Because the WBL is computationally so inexpensive, we can easily calculate
# the DOS and transmission for 10,000 points instead of 1000.
cp $AMSHOME/examples/adf/green_Al/Au.5p .
cp $AMSHOME/examples/adf/green_Al/Au.5p.dirac .
$AMSBIN/dirac < Au.5p.dirac
mv TAPE12 t12.rel
AMS_JOBNAME=Au $AMSBIN/ams <<eor
System
Atoms
Au 0.0 0.0 0.0
End
end
Task SinglePoint
Engine ADF
corepotentials t12.rel
end
create Au file=Au.5p
relativity
level scalar
formalism ZORA
end
xc
lda SCF VWN
end
EndEngine
eor
AMS_JOBNAME=layer $AMSBIN/ams <<eor
System
atoms
Au 0.000000 -4.996959 0.000000
Au 0.000000 -2.498480 -1.442498
Au 0.000000 -2.498480 1.442498
Au 0.000000 0.000000 -2.884996
Au 0.000000 0.000000 0.000000
Au 0.000000 0.000000 2.884996
Au 0.000000 2.498480 -1.442498
Au 0.000000 2.498480 1.442498
Au 0.000000 4.996959 0.000000
end
end
Task SinglePoint
Engine ADF
fragments
Au Au.t21
end
occupations Smearq=0.001
relativity
level scalar
formalism ZORA
end
symmetry NOSYM
title Principal layer
xc
lda SCF VWN
end
EndEngine
eor
AMS_JOBNAME=molecule $AMSBIN/ams <<eor
System
atoms
C -1.400000 0.000000 0.000000
C -0.700000 0.000000 -1.200000
C -0.700000 0.000000 1.200000
C 0.700000 0.000000 -1.200000
C 0.700000 0.000000 1.200000
C 1.400000 0.000000 0.000000
H -1.200000 0.000000 -2.200000
H -1.200000 0.000000 2.200000
H 1.200000 0.000000 -2.200000
H 1.200000 0.000000 2.200000
S -3.200000 0.000000 0.000000
S 3.200000 0.000000 0.000000
end
end
Task SinglePoint
Engine ADF
basis
type DZP
core Large
createOutput None
end
relativity
level scalar
formalism ZORA
end
symmetry NOSYM
title Benzenedithiol
xc
lda SCF VWN
end
EndEngine
eor
AMS_JOBNAME=fock $AMSBIN/ams <<eor
System
atoms
Au -9.911177 -6.662612 0.000000 adf.f=left
Au -9.911178 -4.164133 -1.442498 adf.f=left
Au -9.911178 -4.164133 1.442498 adf.f=left
Au -9.911178 -1.665653 -2.884996 adf.f=left
Au -9.911178 -1.665653 0.000000 adf.f=left
Au -9.911178 -1.665653 2.884996 adf.f=left
Au -9.911178 0.832826 -1.442498 adf.f=left
Au -9.911178 0.832826 1.442498 adf.f=left
Au -9.911178 3.331306 0.000000 adf.f=left
Au -7.555589 -4.996959 0.000000 adf.f=left.2
Au -7.555589 -2.498480 -1.442498 adf.f=left.2
Au -7.555589 -2.498480 1.442498 adf.f=left.2
Au -7.555589 0.000000 -2.884996 adf.f=left.2
Au -7.555589 0.000000 0.000000 adf.f=left.2
Au -7.555589 0.000000 2.884996 adf.f=left.2
Au -7.555589 2.498480 -1.442498 adf.f=left.2
Au -7.555589 2.498480 1.442498 adf.f=left.2
Au -7.555589 4.996959 0.000000 adf.f=left.2
Au -5.200000 -3.331306 0.000000 adf.f=left.3
Au -5.200000 -0.832826 -1.442498 adf.f=left.3
Au -5.200000 -0.832826 1.442498 adf.f=left.3
Au -5.200000 1.665653 -2.884996 adf.f=left.3
Au -5.200000 1.665653 0.000000 adf.f=left.3
Au -5.200000 1.665653 2.884996 adf.f=left.3
Au -5.200000 4.164133 -1.442498 adf.f=left.3
Au -5.200000 4.164133 1.442498 adf.f=left.3
Au -5.200001 6.662612 0.000000 adf.f=left.3
C -1.400000 0.000000 0.000000 adf.f=molecule
C -0.700000 0.000000 -1.200000 adf.f=molecule
C -0.700000 0.000000 1.200000 adf.f=molecule
C 0.700000 0.000000 -1.200000 adf.f=molecule
C 0.700000 0.000000 1.200000 adf.f=molecule
C 1.400000 0.000000 0.000000 adf.f=molecule
H -1.200000 0.000000 -2.200000 adf.f=molecule
H -1.200000 0.000000 2.200000 adf.f=molecule
H 1.200000 0.000000 -2.200000 adf.f=molecule
H 1.200000 0.000000 2.200000 adf.f=molecule
S -3.200000 0.000000 0.000000 adf.f=molecule
S 3.200000 0.000000 0.000000 adf.f=molecule
Au 5.200001 -6.662612 0.000000 adf.f=right.3
Au 5.200000 -4.164133 -1.442498 adf.f=right.3
Au 5.200000 -4.164133 1.442498 adf.f=right.3
Au 5.200000 -1.665653 -2.884996 adf.f=right.3
Au 5.200000 -1.665653 0.000000 adf.f=right.3
Au 5.200000 -1.665653 2.884996 adf.f=right.3
Au 5.200000 0.832826 -1.442498 adf.f=right.3
Au 5.200000 0.832826 1.442498 adf.f=right.3
Au 5.200000 3.331306 0.000000 adf.f=right.3
Au 7.555589 -4.996959 0.000000 adf.f=right.2
Au 7.555589 -2.498480 -1.442498 adf.f=right.2
Au 7.555589 -2.498480 1.442498 adf.f=right.2
Au 7.555589 0.000000 -2.884996 adf.f=right.2
Au 7.555589 0.000000 0.000000 adf.f=right.2
Au 7.555589 0.000000 2.884996 adf.f=right.2
Au 7.555589 2.498480 -1.442498 adf.f=right.2
Au 7.555589 2.498480 1.442498 adf.f=right.2
Au 7.555589 4.996959 0.000000 adf.f=right.2
Au 9.911178 -3.331306 0.000000 adf.f=right
Au 9.911178 -0.832826 -1.442498 adf.f=right
Au 9.911178 -0.832826 1.442498 adf.f=right
Au 9.911178 1.665653 -2.884996 adf.f=right
Au 9.911178 1.665653 0.000000 adf.f=right
Au 9.911178 1.665653 2.884996 adf.f=right
Au 9.911178 4.164133 -1.442498 adf.f=right
Au 9.911178 4.164133 1.442498 adf.f=right
Au 9.911177 6.662612 0.000000 adf.f=right
end
end
Task SinglePoint
Engine ADF
fragments
left layer.t21
left.2 layer.t21
left.3 layer.t21
molecule molecule.t21
right.3 layer.t21
right.2 layer.t21
right layer.t21
end
relativity
level scalar
formalism ZORA
end
symmetry NOSYM
title Benzenedithiol
xc
lda SCF VWN
end
EndEngine
eor
$AMSBIN/green << eor
DOS fock.results/adf.rkf
TRANS fock.results/adf.rkf
EPS -0.5 0 10000
ETA 1e-6
LEFT
FRAGMENT left
ETA 2e-2
END
RIGHT
FRAGMENT right
ETA 2e-2
END
NOSAVE DOS_B, TRANS_B
eor
echo ""
echo "Contents of DOS_A:"
cat DOS_A
echo "END"
echo ""
echo "Contents of TRANS_A:"
cat TRANS_A
echo "END"