Analysis of NMR parameters with Localized Molecular Orbitals¶
Introduction¶
Note
This part requires you to have a license for the NBO6 program. Starting in July 2026, all new AMS2026 and later license files include a license for NBO6. If your AMS2026 or later license file does not include NBO6, please request a new license by contacting license@scm.com.
Localized molecular orbitals, such as Natural Bonding Orbitals (NBO) or Natural Localized Molecular Orbitals (NLMO) can be helpful in analyzing and interpreting properties computed in electronic structure calculations.
This tutorial analyzes the effects of NH2 and NO2 substituents in ortho-, meta-, and para-positions on NMR chemical shifts of aromatic carbon atoms in a benzene ring. These shifts in aromatic systems are commonly believed to originate from a depletion or accumulation of electrons in the π-orbitals at these atoms. We examine this claim by means of NMR calculations with ADF and NBO/NLMO analysis tools.
This tutorial loosely follows the following publication: R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach The unexpected roles of σ and π orbitals in electron donor and acceptor group effects on the 13C NMR chemical shifts in substituted benzenes, Chem. Sci. Phys. 8, 6570-6576 (2017).
Step 1: Preparations¶
The molecular structures needed for this tutorial can be downloaded by clicking on the following links:
These structures have already been optimized and properly aligned. In general, you should optimize the structures first.
Note
The analysis of NMR and EFG results in terms of localized molecular orbitals requires some care about the alignment of the molecules when comparing results between different systems as in our case. This is, for example, because the analysis in terms of NLMOs is origin dependent. To obtain comparable orbital contributions one has furthermore to ensure that the different molecules are oriented in the same way. In AMSinput you can use the options Edit → Align and Edit → Set Origin to achieve this when performing localized orbital analysis with your custom models.
The main idea is now to compute the NMR shielding parameters for three different carbon atoms in the ring, C(1), C(4), and C(5), which are in ortho-, meta-, and para-position to the substituent at atom C(3):
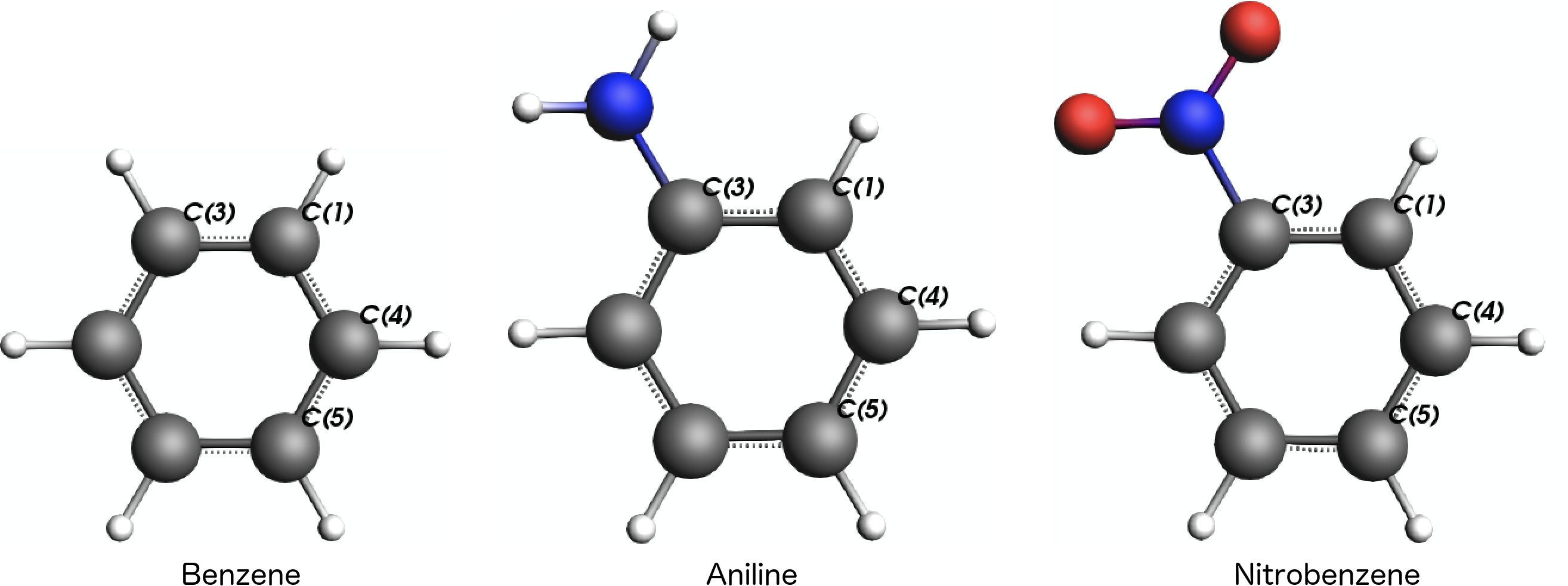
Step 2: Calculation Settings¶
We need to perform three ADF calculations, one for each molecule (i.e. Benzene, Aniline, and Nitrobenzene).
Let us start with Benzene:
Next, select the following settings in the main AMSinput panel
Note
An all-electron basis set is desirable because a hybrid functional is used. Using an all-electron basis set also improves the quality of the NMR results.
Note
A scalar relativistic treatment (Relativity → Scalar) is required to perform the NBO analysis of the NMR shifts.
In order to compare the computed NMR results with their experimental counterparts (which were measured in Chloroform solution), we enable the COSMO solvation model:
We now select the atoms for which we want to compute the NMR properties:

and enable the NBO analysis of the NMR properties:
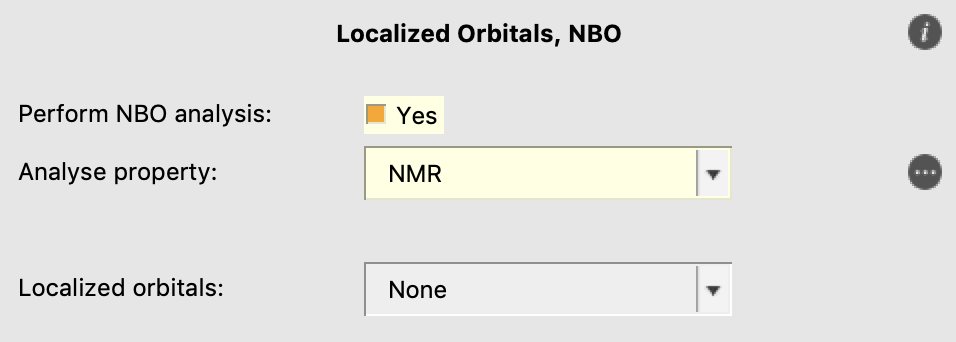
Finally, we need to disable the automatic symmetry treatment of ADF:
Now that all options are set, save the job:
BenzeneFor the other two systems (i.e. Aniline, and Nitrobenzene), we can avoid having to set again most computational options:
Aniline.xyz and click OpenAnilineNitrobenzene.xyzStep 3: Running the Calculations¶
We are now ready to run all three calculations
NMR Results¶
After all three calculations are completed (this can take 10 minutes up to several hours, depending on your hardware), we can explore the output file:
In the AMSoutput window, we can see the output file. We can search for text on it by using the text box localized at the bottom. For example, we can go directly to the NMR section by searching the text N M R, or go to the beginning of the shielding tensor calculation section for each atom with **** N U C L E U S :. Notice that in the latter case, there is one section for each of the three nuclei C(1), C(4), and C(5), and you can reach each one of them by using the small triangles to the right of the searching text box.
We can also retrieve the isotropic shielding constants directly by clicking on the properties listed on the top menu of AMSoutput:
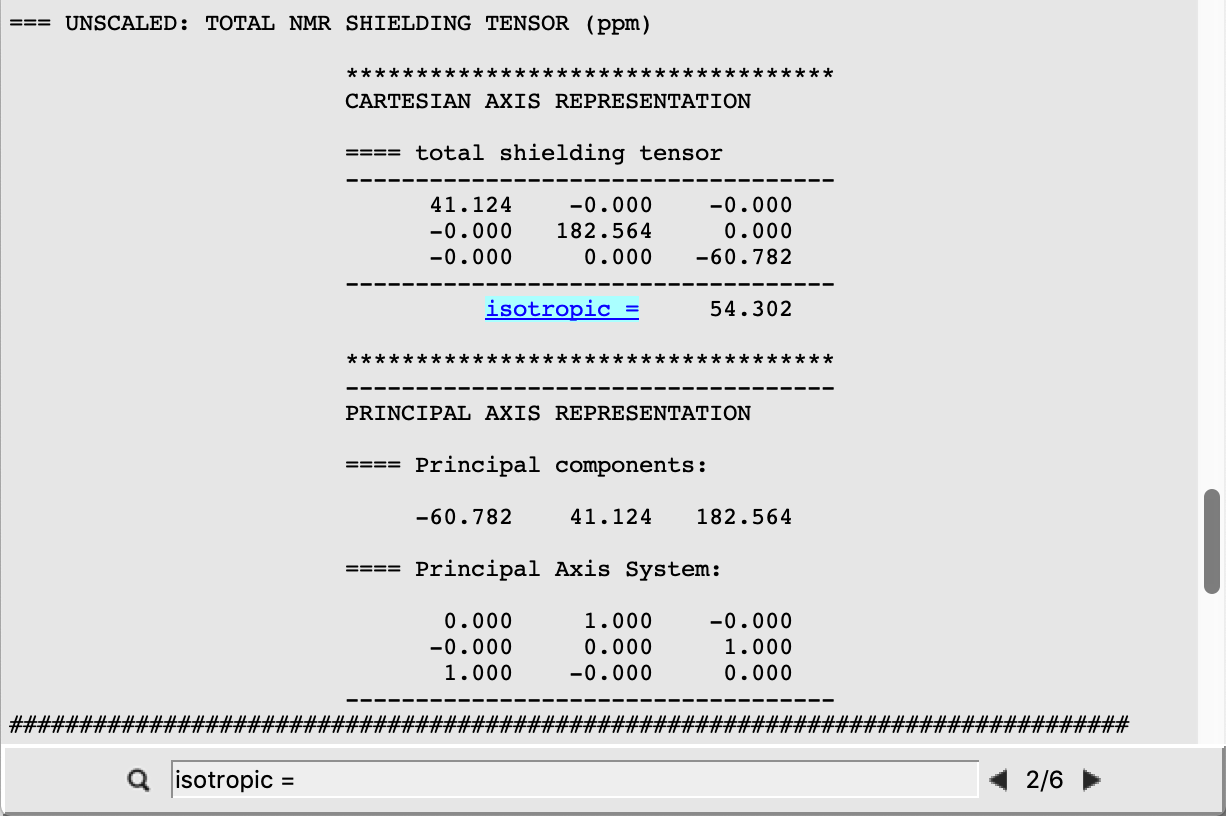
Repeating the previous click, we can retrieve the result for each atom.
In the case of Benzene the atoms C(1), C(4), and C(5) are all equivalent, thus yielding shielding isotropic constants σi of the same value. This is different in the case of the other two systems, whose shielding constants are retrieved analogously from the corresponding output files. The following are the expected results:
Compound |
Substitution |
σi (ppm) |
|---|---|---|
Benzene |
54.302 |
|
Aniline |
o-NH2 |
69.391 |
m-NH2 |
53.198 |
|
p-NH2 |
66.490 |
|
Nitrobenzene |
o-NO2 |
57.630 |
m-NO2 |
53.927 |
|
p-NO2 |
46.071 |
The chemical shifts δi are then computed relative to the unsubstituted Benzene molecule as follows:
δi = σBenzene - σi + δBenzene
Here σBenzene is the isotropic value of the chemical shielding computed σi for Benzene (54.302 ppm in our case), and δBenzene = 128.55 ppm is the experimental chemical shift relative to TMS for 13C in Benzene.
Note
It is important to note that the trends analyzed herein are not affected by the nature of the intermediate reference.
This yields the following results:
Compound |
Substitution |
exp 13C-shift |
calc 13C-shift |
|---|---|---|---|
Benzene |
128.55 |
128.55 (by definition) |
|
Aniline |
o-NH2 |
115.29 |
113.46 |
m-NH2 |
129.45 |
129.65 |
|
p-NH2 |
118.73 |
116.36 |
|
Nitrobenzene |
o-NO2 |
123.65 |
125.22 |
m-NO2 |
129.48 |
128.92 |
|
p-NO2 |
134.76 |
136.78 |
The experimental NMR shifts are reproduced with an error margin of about 2 ppm, and the trend between the different substitutions is reproduced: the NH2 group leads to a significant decrease in the chemical shifts in ortho- and para- position. The meta-position remains almost unaffected by the NH2 group. This is also observed for the meta-position near a NO2 substitution, while the chemical shifts are lowered at the ortho-atom and increased at the para-position.
To rationalize this finding, we proceed to examine the contributions of individual orbitals to these substituent effects.
NLMO/NBO Analysis¶
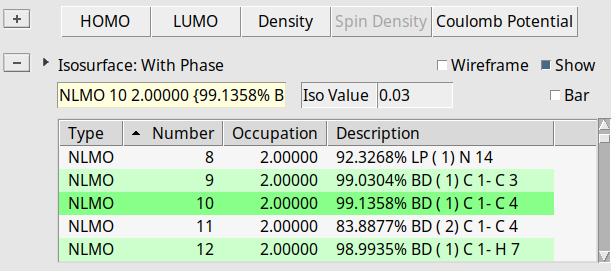
We first look at the individual NLMO contributions to the isotropic shielding tensors. Each output file contains the NLMO and NBO decomposition of the isotropic shielding tensors (and other quantities) for the atoms C(1), C(4), and C(5).
The NMR shielding tensors consist of diamagnetic and paramagnetic (+SO) contributions. Diamagnetic terms are larger but mostly dominated by contributions from core orbitals and they are essentially not affected by the local environment of an atom. As we are interested in substitution effects, i.e. changing environments around an atom, we focus on the diamagnetic terms here as these are most influential on the magnitude and direction of the chemical shifts.
NLMO contributions to in the search bar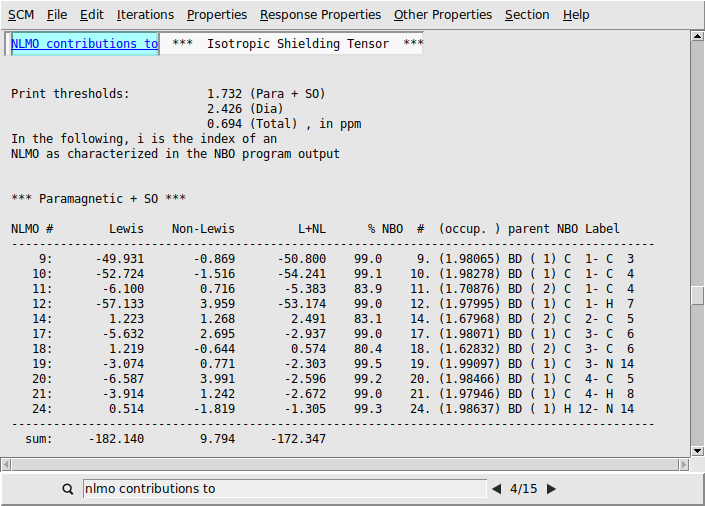
We find that the paramagnetic part of the isotropic shielding tensor at atom C(1) is mostly determined by contributions originating from three bonding NLMOs formed between C(1) and its three neighbors C(3), C(4), and H(7). See labels C 1- C 3, C 1- C 4, and C 1- H 7 respectively.
Analogous results are obtained for C(4) and C(5) as well as for the corresponding atoms in the Nitrobenzene molecule.
One finds similar results when examining the NBO contributions to the isotropic shielding tensors.
These contributions can be obtained by searching for NBO contributions to in the search bar of ADFOutput.
Note
The following table summarizes characteristics of the common NBO/NLMO types, showing the number of centers, quantum shell, Lewis(L)/non-Lewis(NL) donor-acceptor type, and the program output label:
NBO Type |
Centers |
Shell |
L/NL |
Label |
|---|---|---|---|---|
core |
1-c |
core |
L |
CR |
nonbonded (lone pair) |
1-c |
valence |
L |
LP |
bond |
2-c |
valence |
L |
BD |
antibond |
2-c |
valence |
NL |
BD* |
Rydberg |
1-c |
Rydberg |
NL |
RY |
Inspecting NLMOs¶
We now examine in more detail the NLMOs with the largest contributions.
The selection window allows you to pick individual orbitals for plotting in AMSview
A visual inspection of the three NLMOs with the largest contribution to the isotropic shielding tensor reveals that they all have a pronounced σ-orbital character:
0.06 as isovalue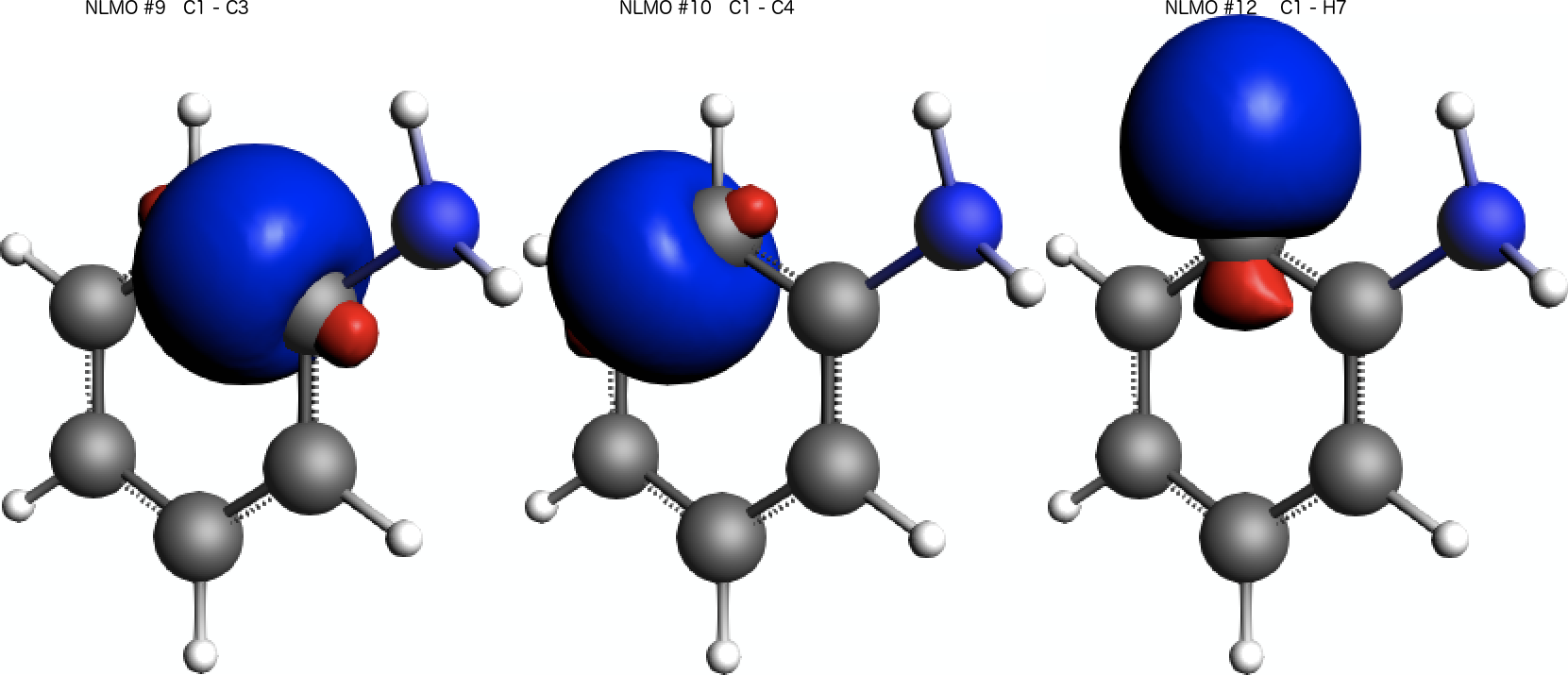
Opposed to that, the π-orbitals of the aromatic system can be clearly identified as well, e.g. for NLMO #11 in the above listing of contributions
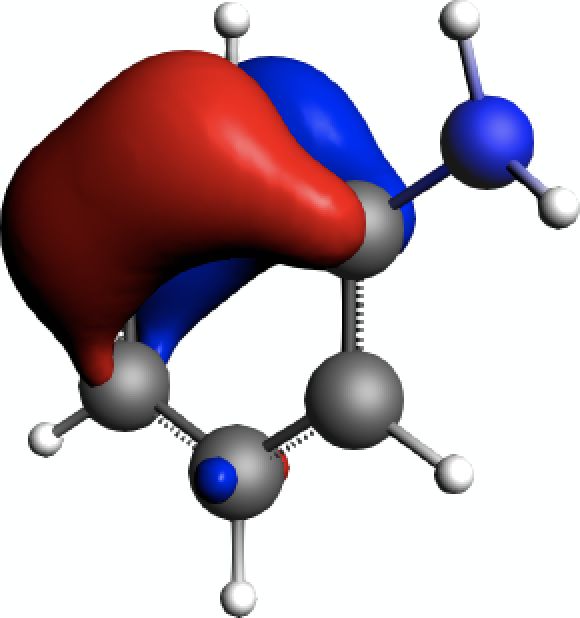
As the results discussed above show, such π-orbitals are not contributing much to the isotropic shielding tensors.
The corresponding analysis in terms of NBOs can be done in exactly the same way
and suggests exactly the same finding
In conclusion, the chemical shifts due to substitutions on an aromatic ring are mainly influenced by contributions from σ-bonding orbitals rather than π-orbitals.
Further Reading¶
R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach The unexpected roles of σ and π orbitals in electron donor and acceptor group effects on the 13C NMR chemical shifts in substituted benzenes, Chem. Sci. Phys. 8, 6570-6576 (2017).
J. A. Bohmann, F. Weinhold and T. C. Farrar Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations, J. Chem. Phys. 107, 1173-1184 (1997).
J. Autschbach Analyzing NMR shielding tensors calculated with two-component relativistic methods using spin-free localized molecular orbitals, J. Chem. Phys. 128, 164112 (2008).
R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach The halogen effect on the 13C NMR chemical shift in substituted benzenes, Phys. Chem. Chem. Phys. 20, 11247 (2018).