4. Command Line Tool¶
The two command-line tools are run_acerxn.py and setup_acerxn.py. The former runs an ACErxn job, and the latter creates a full input file for run_acerxn.py from minimal input information. They both expect a text input file in the general AMS block format.
4.1. System information¶
The most important part of the input is the molecular information about the reactants and the products. They need to be provided as AMS system blocks. Whether the system block represents a reactant molecule, a product molecule, or anything else is determined by the block title.
4.1.1. Reactant System Blocks¶
The most elaborate system blocks are those for reactant molecules, as they will be used to determine much of the information needed by the ACErxn process. ACErxn needs to know the following:
What the smallest possible fragments of the system are. The bonds between the atoms in these fragments will never be broken.
The charges of these smallest possible fragments.
The stability of these smallest possible fragments. If a fragment is determined to be unstable, it will be required to undergo change in every elementary reaction of the network.
The above information can be guessed automatically by the ACErxn code if the following information is provided by the user.
Which atoms in the reactant molecules are active atoms (can partake in bond breaking/forming).
The charge of the reactant molecules.
If there are any bonds between active atoms that can never be broken.
This minimal information can be used to directly run ACErxn via the ‘run_acerxn.py’ script, or the setup_acerxn.py script can be used to check the guesses the program made for the fragments.
The system blocks mostly behave as general AMS system blocks, with all the available sections. A reactant charge can be provided, and if no charge is provided, it will be assumed zero. Bonds and bond orders can be provided, and if they are not, they will be guessed using the PLAMS routines.
There is also information that is very specific to ACErxn in the system block. For instance, for each atom in a reactant molecule an entry is needed specifying whether it is an active atom. An active atom is an atom that can partake in bond breaking/forming.
The title of the reactant system block needs to start with the word ‘reactant’. The integer following is arbitrary, but needs to be unique.
System reactant0
Atoms
O -1.569978734409354 -5.334723658758318 -1.768979030791651e-15 active=True
H -1.465525886811915 -6.114331990252619 0.5503639269551164 active=True
H -1.88333727720167 -4.603418122596448 0.5372137716385108 active=False
End
BondOrders
2 1 1.0
3 1 1.0
End
Charge 0
End
In some cases, two atoms need to be designated with an ‘active’ label, even though the bond between them should never break. An example would be a dihydroxy-alkane, where the C-OH bonds can be broken, and therefore all C and O atoms must be active. However, the C-C bond is expected to be stable. In that case, the carbon-carbon bond can be specified as frozen in the system block.
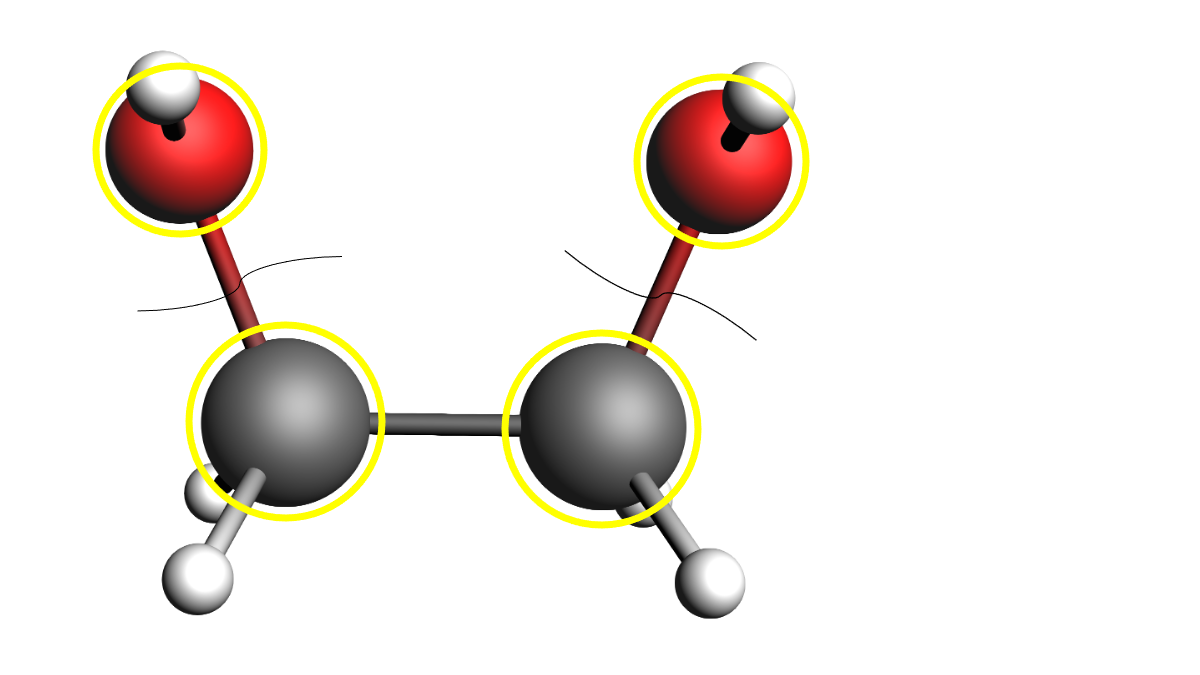
System
Atoms
C -2.416513737568649 -2.36446827946751 0.000416972679169055 active=True
C -0.8777937308441511 -2.288433140361173 0.001290215051439407 active=True
O -0.2886240156882122 -3.562466941373763 -0.0086490566181646 active=True
O -2.877943398843959 -3.690109811405948 -0.007739169128441394 active=True
H -2.807563695755317 -1.855220536347314 -0.9063168955577124 active=False
H -2.825707514338756 -1.814970611037596 0.8798905235190039 active=False
H -0.5255263895182007 -1.702468329618303 0.8818578162320893 active=False
H -0.5371656009354311 -1.741027777693658 -0.903575452997571 active=False
H -0.2445864773000304 -3.863161081612486 0.9365262244532915 active=False
H -2.879667871430366 -3.99694614875279 0.9364921818555469 active=False
End
BondOrders
1 2 1.0 frozen=True
1 4 1.0
1 5 1.0
1 6 1.0
2 3 1.0
2 7 1.0
2 8 1.0
3 9 1.0
4 10 1.0
End
End
When running the ‘setup_acerxn.py’ script on the above reactant system blocks, a new system block is written that contains all fragment information. You can then adjust this information to your liking and pass the full system block to the ‘run_acerxn.py’ script.
system reactant0
Atoms
O 0.3133585428 0.7796083315 0.0000000000 active=True region=frag0
H 0.4178113904 0.0000000000 0.5503639270 active=True region=frag1
H 0.0000000000 1.5109138677 0.5372137716 active=False region=frag0
End
BondOrders
2 1 1.0
3 1 1.0
End
Charge 0.0
Region frag0
Properties
Charge -1.0
Stable Yes
End
End
Region frag1
Properties
Charge 1.0
Stable No
End
End
End
4.1.2. Other System Blocks¶
If the molecule is a product, only regular AMS system information is needed. If a charge is provided, it will be ignored by the code, since the product molecule charges are estimated from the charges of the smallest fragments. The title of the product system block needs to start with the word ‘product’, followed by a unique integer.
System product0
Atoms
O -1.569978734409354 -5.334723658758318 -1.768979030791651e-15
H -1.465525886811915 -6.114331990252619 0.5503639269551164
End
BondOrders
2 1 1.0
End
End
Optionally, the user can provide intermediates. ACErxn will then attempt to find reaction paths that include all these intermediates. We define a single intermediate as a stable (set of) molecule(s) with the same stoichiometry as the combined reactant molecules. As for product system blocks, only regular AMS system information is needed, and if a charge is provided it will be ignored by the code. You can provide many intermediates and define a system block for each intermediate submolecule. The name of the system block needs to start with ‘IM’, followed by a number unique to the intermediate. This should then be followed by an underscore, and another integer describing which submolecule of the intermediate this system block represents.
System IM0_0
Atoms
C -3.24062111284074 -1.445184670722061 0.0009376243020079313
C -1.700202188513091 -1.436988320050662 -0.001542452608866306
H -3.7630958635082 -3.057071318965873 0.9409792289436294
O -3.759682499867571 -2.749001444990656 -0.00308032012251173
H -3.610856684448746 -0.9206380903104889 -0.9056146436732758
H -3.622899214012238 -0.8779711360102836 0.8807988470236872
H -1.320135077567553 -0.864620543066376 0.8754671468246595
H -1.337920744004371 -0.9116253182098165 -0.9108129286311496
End
BondOrders
1 2 1.0
1 4 1.0
1 5 1.0
1 6 1.0
2 7 1.0
2 8 1.0
4 3 1.0
End
End
System IM0_1
Atoms
O -1.569978734409354 -5.334723658758318 -1.768979030791651e-15
H -1.465525886811915 -6.114331990252619 0.5503639269551164
End
BondOrders
2 1 1.0
End
End
It is also possible to provide system blocks for known single molecules and forbidden molecules. The former should have the name ‘mol’ followed by an integer, and the latter should have the name ‘forbidden’ followed by an integer.
4.2. Settings information¶
The settings need to be provided in general AMS format. The keywords are described in the next section, with only a short example shown here.
RunInfo
Steps GenerateIntermediates
End
4.3. Engine information¶
For the QM geometry optimizations in the first step of ACErxn (intermediate generation), an engine block needs to be provided in general AMS format. Optionally, an additional engine block can be provided for a ForceField engine, which will then be used for the initial geometry optimization. If not provided, UFF with default settings will be used. Currently UFF is the only engine that can be selected for the initial optimization.
Engine Mopac
Model PM6
EndEngine
Engine ForceField
Type UFF
UFF
Library UFF4MOF
End
EndEngine
4.4. Summary¶
The following minimal input file can be run with both the scripts ‘setup_acerxn.py’ and ‘run_acerxn.py’.
$AMSBIN/amspython $AMSHOME/scripting/scm/acerxn/setup_acerxn.py << eor
system reactant0
Atoms
C 1.0700000000 2.4608000000 0.8738000000 active=True
H 1.4267000000 2.9652000000 0.0001000000 active=False
H 0.0000000000 2.4608000000 0.8738000000 active=False
C 1.5833000000 1.0088000000 0.8738000000 active=False
H 1.2266000000 0.5044000000 1.7474000000 active=False
H 1.2266000000 0.5044000000 0.0001000000 active=False
C 3.1233000000 1.0088000000 0.8738000000 active=False
H 3.4800000000 0.0000000000 0.8739000000 active=False
H 3.4800000000 1.5130000000 0.0000000000 active=False
C 3.6367000000 1.7350000000 2.1310000000 active=False
H 4.6201000000 1.3862000000 2.3678000000 active=False
H 3.6666000000 2.7887000000 1.9471000000 active=False
O 2.7590000000 1.4674000000 3.2278000000 active=True
N 3.4288000000 1.6425000000 4.3984000000 active=True
O 4.0183000000 1.7966000000 5.4285000000 active=False
H 1.4267000000 2.9652000000 1.7474000000 active=True
End
BondOrders
10 11 1
1 16 1
4 5 1
7 10 1
13 14 1
1 3 1
4 7 1
7 8 1
10 13 1
7 9 1
1 4 1
10 12 1
1 2 1
4 6 1
14 15 2.0
End
Charge 0
End
system product0
Atoms
C -2.6271620000 2.3237140000 0.0648140000
H -2.2704890000 2.8281120000 -0.8088370000
H -3.6971620000 2.3237270000 0.0648140000
C -2.1138200000 3.0496700000 1.3222190000
H -2.4704580000 4.0584860000 1.3222100000
H -1.0438200000 3.0496400000 1.3222290000
C -2.6271850000 2.3237290000 2.5796230000
H -2.2705100000 2.8281250000 3.4532750000
H -3.6971850000 2.3237610000 2.5796150000
C -2.1138950000 0.8717870000 2.5796340000
H -2.0839500000 0.5040910000 3.5840260000
H -1.1304640000 0.8411400000 2.1591300000
O -3.0307730000 0.0154900000 1.8788390000
N -2.0980060000 0.9782090000 -0.0147070000
O -1.5860940000 0.5028570000 -1.0362940000
H -3.6277380000 -0.4600880000 2.4611260000
End
BondOrders
1 2 1
10 12 1
4 7 1
1 4 1
13 16 1
1 3 1
4 6 1
7 9 1
7 8 1
10 11 1
4 5 1
7 10 1
10 13 1
1 14 1
14 15 2.0
End
End
RunInfo
Steps GenerateIntermediates
End
BasicOptions MaxJobs 4
Engine Mopac
Model PM6
EndEngine
eor
The script setup.py returns an output file that can be adjusted and then used as an input file for ‘run_acerxn.py’
$AMSBIN/amspython $AMSHOME/scripting/scm/acerxn/run_acerxn.py << eor
system reactant0
Atoms
C 1.0700000000 2.4608000000 0.8738000000 active=True region=frag0
H 1.4267000000 2.9652000000 0.0001000000 active=False region=frag0
H 0.0000000000 2.4608000000 0.8738000000 active=False region=frag0
C 1.5833000000 1.0088000000 0.8738000000 active=False region=frag0
H 1.2266000000 0.5044000000 1.7474000000 active=False region=frag0
H 1.2266000000 0.5044000000 0.0001000000 active=False region=frag0
C 3.1233000000 1.0088000000 0.8738000000 active=False region=frag0
H 3.4800000000 0.0000000000 0.8739000000 active=False region=frag0
H 3.4800000000 1.5130000000 0.0000000000 active=False region=frag0
C 3.6367000000 1.7350000000 2.1310000000 active=False region=frag0
H 4.6201000000 1.3862000000 2.3678000000 active=False region=frag0
H 3.6666000000 2.7887000000 1.9471000000 active=False region=frag0
O 2.7590000000 1.4674000000 3.2278000000 active=True region=frag0
N 3.4288000000 1.6425000000 4.3984000000 active=True region=frag1
O 4.0183000000 1.7966000000 5.4285000000 active=False region=frag1
H 1.4267000000 2.9652000000 1.7474000000 active=True region=frag2
End
BondOrders
10 11 1.0
1 16 1.0
4 5 1.0
7 10 1.0
13 14 1.0
1 3 1.0
4 7 1.0
7 8 1.0
10 13 1.0
7 9 1.0
1 4 1.0
10 12 1.0
1 2 1.0
4 6 1.0
14 15 2.0
End
Charge 0.0
Region frag0
Properties
Charge -1.0
Stable No
End
End
Region frag1
Properties
Charge 0.0
Stable Yes
End
End
Region frag2
Properties
Charge 1.0
Stable No
End
End
End
system product0_0
Atoms
C -2.6271620000 2.3237140000 0.0648140000
H -2.2704890000 2.8281120000 -0.8088370000
H -3.6971620000 2.3237270000 0.0648140000
C -2.1138200000 3.0496700000 1.3222190000
H -2.4704580000 4.0584860000 1.3222100000
H -1.0438200000 3.0496400000 1.3222290000
C -2.6271850000 2.3237290000 2.5796230000
H -2.2705100000 2.8281250000 3.4532750000
H -3.6971850000 2.3237610000 2.5796150000
C -2.1138950000 0.8717870000 2.5796340000
H -2.0839500000 0.5040910000 3.5840260000
H -1.1304640000 0.8411400000 2.1591300000
O -3.0307730000 0.0154900000 1.8788390000
N -2.0980060000 0.9782090000 -0.0147070000
O -1.5860940000 0.5028570000 -1.0362940000
H -3.6277380000 -0.4600880000 2.4611260000
End
BondOrders
1 2 1.0
10 12 1.0
4 7 1.0
1 4 1.0
13 16 1.0
1 3 1.0
4 6 1.0
7 9 1.0
7 8 1.0
10 11 1.0
4 5 1.0
7 10 1.0
10 13 1.0
1 14 1.0
14 15 2.0
End
End
RunInfo
Steps GenerateIntermediates
End
BasicOptions MaxJobs 4
Engine mopac
model PM6
EndEngine