Active Learning: Single-Molecule Production Simulation¶
Reuse the retrained active-learning model in a production calculation, then inspect optimized structures and vibrational properties for the single-molecule test case.
Initial imports and load active learning job from disk¶
from scm.simple_active_learning import SimpleActiveLearningJob
import scm.plams as plams
import os
import matplotlib.pyplot as plt
plams.init()
PLAMS working folder: /path/to/plams_workdir.003
# replace the path with your own path !
previous_sal_job_path = os.path.expandvars(
"$AMSHOME/examples/SAL/Output/SingleMolecule/plams_workdir/sal"
)
sal_job = SimpleActiveLearningJob.load_external(previous_sal_job_path)
params_job = sal_job.results.get_params_job()
Structure for production job¶
We could initialize a PLAMS molecule in many different ways. Here, we get the final frame from the final production simulation in the SAL job, and preoptimize it with UFF.
molecule = sal_job.results.get_main_molecule()
molecule = plams.preoptimize(molecule, model="UFF")
plams.plot_molecule(molecule)

Settings for production job¶
Let’s run a geometry optimization + frequencies. But you could run any AMS job!
The MaxRestarts option is useful when calculating normal modes. If the geometry optimizer converges to a transition state, it will continue until it finds a local minimum!
s = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.Properties.NormalModes = "Yes"
s.input.ams.GeometryOptimization.MaxRestarts = 5
s.runscript.nproc = 1
retrained_engine_settings = params_job.results.get_production_engine_settings()
new_job = plams.AMSJob(
settings=s + retrained_engine_settings, name="retrained_m3gnet", molecule=molecule
)
print(new_job.get_input())
GeometryOptimization
MaxRestarts 5
End
Properties
... output trimmed ....
Model Custom
ParameterDir /path/to/plams_workdir/sal/step4_attempt1_training/results/optimization/m3gnet/m3gnet
EndEngine
new_job.run();
[02.02|12:39:32] JOB retrained_m3gnet STARTED
[02.02|12:39:32] JOB retrained_m3gnet RUNNING
[02.02|12:39:47] JOB retrained_m3gnet FINISHED
[02.02|12:39:47] JOB retrained_m3gnet SUCCESSFUL
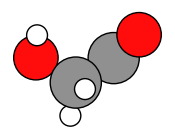
Optimized structure¶
plams.plot_molecule(new_job.results.get_main_molecule())
Frequencies¶
width = 50 # cm^-1
x, y = new_job.results.get_ir_spectrum(
broadening_width=width, post_process="all_intensities_to_1"
)
plt.plot(x, y, label="Retrained M3GNet")
plt.xlabel("Frequency (cm^-1)")
plt.ylabel("Normal mode count")
plt.legend();

Compare to reference UFF¶
In this case, we can also calculate the normal modes with the reference method (UFF) and compare:
ref_engine_settings = plams.Settings()
ref_engine_settings.input.ForceField.Type = "UFF"
ref_job = plams.AMSJob(
settings=s + ref_engine_settings, name="uff_ref", molecule=molecule
)
ref_job.run();
[02.02|12:39:48] JOB uff_ref STARTED
[02.02|12:39:48] JOB uff_ref RUNNING
[02.02|12:39:48] JOB uff_ref FINISHED
[02.02|12:39:48] JOB uff_ref SUCCESSFUL
retrained_structure_rmsd = plams.Molecule.rmsd(
ref_job.results.get_main_molecule(),
new_job.results.get_main_molecule(),
ignore_hydrogen=True,
)
print(f"Structural RMSD: {retrained_structure_rmsd:.2f} angstrom")
Structural RMSD: 0.10 angstrom
x_ref, y_ref = ref_job.results.get_ir_spectrum(
broadening_width=width, post_process="all_intensities_to_1"
)
plt.plot(x, y, label="Retrained M3GNet")
plt.plot(x_ref, y_ref, label="UFF (reference method)")
plt.xlabel("Frequency (cm^-1)")
plt.ylabel("Normal mode count")
plt.legend();

The agreement looks very good! The only significant difference is the highest-frequency vibration (the O-H streching vibration). This frequency is very sensitive to the calculated forces near the equilibrium (minimum) structure. The agreement could have been improved by
having more training data, for example by setting a tighter success criterion for the forces and energy in the active learning
running the active learning MD at a lower temperature (closer to the equilibrium structure, but this would mean less conformational sampling)
training for more epochs
Tip: check if the vibrational frequencies with retrained M3GNet or M3GNet-UP-2022 agree better or worse with the reference UFF calculation.
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial imports and load active learning job from disk
from scm.simple_active_learning import SimpleActiveLearningJob
import scm.plams as plams
import os
import matplotlib.pyplot as plt
plams.init()
# replace the path with your own path !
previous_sal_job_path = os.path.expandvars("$AMSHOME/examples/SAL/Output/SingleMolecule/plams_workdir/sal")
sal_job = SimpleActiveLearningJob.load_external(previous_sal_job_path)
params_job = sal_job.results.get_params_job()
# ## Structure for production job
#
# We could initialize a PLAMS molecule in many different ways. Here, we get the final frame from the final production simulation in the SAL job, and preoptimize it with UFF.
molecule = sal_job.results.get_main_molecule()
molecule = plams.preoptimize(molecule, model="UFF")
plams.plot_molecule(molecule)
# ## Settings for production job
#
# Let's run a geometry optimization + frequencies. But you could run any AMS job!
#
# The ``MaxRestarts`` option is useful when calculating normal modes. If the geometry optimizer converges to a transition state, it will continue until it finds a local minimum!
s = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.Properties.NormalModes = "Yes"
s.input.ams.GeometryOptimization.MaxRestarts = 5
s.runscript.nproc = 1
retrained_engine_settings = params_job.results.get_production_engine_settings()
new_job = plams.AMSJob(settings=s + retrained_engine_settings, name="retrained_m3gnet", molecule=molecule)
print(new_job.get_input())
new_job.run()
# ### Optimized structure
plams.plot_molecule(new_job.results.get_main_molecule())
# ### Frequencies
width = 50 # cm^-1
x, y = new_job.results.get_ir_spectrum(broadening_width=width, post_process="all_intensities_to_1")
plt.plot(x, y, label="Retrained M3GNet")
plt.xlabel("Frequency (cm^-1)")
plt.ylabel("Normal mode count")
plt.legend()
plt.gcf().savefig("picture1.png")
plt.clf()
# ## Compare to reference UFF
# In this case, we can also calculate the normal modes with the reference method (UFF) and compare:
ref_engine_settings = plams.Settings()
ref_engine_settings.input.ForceField.Type = "UFF"
ref_job = plams.AMSJob(settings=s + ref_engine_settings, name="uff_ref", molecule=molecule)
ref_job.run()
retrained_structure_rmsd = plams.Molecule.rmsd(
ref_job.results.get_main_molecule(),
new_job.results.get_main_molecule(),
ignore_hydrogen=True,
)
print(f"Structural RMSD: {retrained_structure_rmsd:.2f} angstrom")
x_ref, y_ref = ref_job.results.get_ir_spectrum(broadening_width=width, post_process="all_intensities_to_1")
plt.plot(x, y, label="Retrained M3GNet")
plt.plot(x_ref, y_ref, label="UFF (reference method)")
plt.xlabel("Frequency (cm^-1)")
plt.ylabel("Normal mode count")
plt.legend()
plt.gcf().savefig("picture2.png")
plt.clf()
# The agreement looks very good! The only significant difference is the highest-frequency vibration (the O-H streching vibration). This frequency is very sensitive to the calculated forces near the equilibrium (minimum) structure. The agreement could have been improved by
#
# * having more training data, for example by setting a tighter success criterion for the forces and energy in the active learning
# * running the active learning MD at a lower temperature (closer to the equilibrium structure, but this would mean less conformational sampling)
# * training for more epochs
#
# Tip: check if the vibrational frequencies with retrained M3GNet or M3GNet-UP-2022 agree better or worse with the reference UFF calculation.