Reaction Discovery with ReaxFF¶
This example illustrates how to use Reactions Discovery with AMS. See also the Reactions Discovery documentation.
The example asks what products may form from NH2-CH2-CH2-OH + CO2 + H2O and shows how side products such as NH3, NH2-CH2-CH=O, and OH-NH-CH2-CH2-OH can emerge from the exploration.
Because Reactions Discovery starts from random packed configurations, repeated runs will not produce exactly the same set of products.
Initial imports¶
from typing import List
import scm.plams as plams
from scm.input_classes import engines
from scm.reactions_discovery import ReactionsDiscoveryJob
from rdkit import Chem
from rdkit.Chem import Draw
# Settings for displaying molecules in the notebook
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.ipython_useSVG = True
IPythonConsole.molSize = 250, 250
try:
from scm.plams import view # view molecule using AMSview in a Jupyter Notebook in AMS2026+
except ImportError:
from scm.plams import plot_molecule # plot molecule in a Jupyter Notebook in AMS2023+
def view(molecule, **kwargs):
plot_molecule(molecule)
# this line is not required in AMS2025+
plams.init()
Helpers for showing molecules¶
def draw_molecules(molecules: List[plams.Molecule]):
smiles = [molecule.properties.smiles for molecule in molecules]
return draw_smiles(smiles)
def draw_smiles(smiles: List[str]):
rd_mols = [Chem.MolFromSmiles(s) for s in smiles]
return Draw.MolsToGridImage(rd_mols)
The ReactionsDiscoveryJob class¶
job = ReactionsDiscoveryJob(name="MyDiscovery")
driver = job.input
md = driver.MolecularDynamics
Setting up the reactants for molecular dynamics¶
md.NumSimulations = 4
build = md.BuildSystem
build.NumAtoms = 250
build.Density = 0.9
build.Molecule[0].SMILES = "O" # Water
build.Molecule[0].MoleFraction = 1
build.Molecule[1].SMILES = "NCCO" # MEA
build.Molecule[1].MoleFraction = 2
build.Molecule[2].SMILES = "O=C=O" # Carbondioxide
build.Molecule[2].MoleFraction = 3
draw_smiles([build.Molecule[i].SMILES.val for i in range(len(build.Molecule))])

Setting up reactive molecular dynamics¶
md.Enabled = "Yes"
md.Type = "NanoReactor"
reactor = md.NanoReactor
reactor.NumCycles = 10
reactor.Temperature = 500
reactor.MinVolumeFraction = 0.6
Setting up network extraction and ranking¶
network = driver.NetworkExtraction
network.Enabled = "Yes"
network.UseCharges = "Yes"
ranking = driver.ProductRanking
ranking.Enabled = "Yes"
Selecting the AMS engine to use¶
engine = engines.ReaxFF()
engine.ForceField = "Glycine.ff"
engine.TaperBO = (
"Yes" # This is a really important setting for reaction analysis with ReaxFF potentials
)
driver.Engine = engine
Running reactions discovery¶
result = job.run() # start the job
job.check() # check if job was successful
[13.01|09:49:52] JOB MyDiscovery STARTED
[13.01|09:49:52] JOB MyDiscovery RUNNING
[13.01|10:26:59] JOB MyDiscovery FINISHED
[13.01|10:27:00] Job mdsim_2 reported warnings. Please check the output
[13.01|10:27:00] JOB MyDiscovery SUCCESSFUL
[13.01|10:27:00] Job mdsim_2 reported warnings. Please check the output
True
Obtain the results¶
graph, molecules, categories = result.get_network()
Categories¶
The categories are Products Reactants and Unstable, as
described in the reactions discovery manual. molecules is a
dictionary with keys equal to the categories and each concomitant value
is a list of PLAMS molecules.
print(categories)
['Products', 'Reactants', 'Unstable']
draw_molecules(molecules["Reactants"])

Products¶
These are the side products that reactions discovery found in the order as found by the ranking algorithm.
draw_molecules(molecules["Products"][:6])

Unstable¶
Unstable products were determined to not likely exist outside of reactive dynamics. This e.g. includes radicals or structures that don’t form stable molecules in isolation. Not all unstable molecules have a sensible 2d structure, so instead we plot their 3d structure.
view(molecules["Unstable"][0], width=200, height=200)
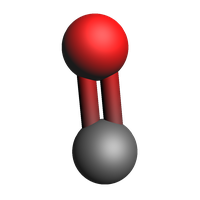
view(molecules["Unstable"][1], width=200, height=200)
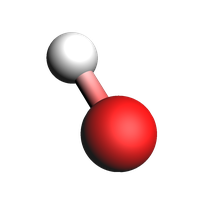
view(molecules["Unstable"][2], width=200, height=200)
Graph of the reaction network¶
The graph is a bipartate networkx DiGraph with reaction and molecule
nodes. This can be stored on disk in standard graph formats,
e.g. .gml
import networkx as nx
nx.write_gml(graph, "reaction_network.gml")
Load a job not originally run by PLAMS¶
from scm.plams import FileError
try:
job = ReactionsDiscoveryJob.load_external("plams_workdir/MyDiscovery")
graph, molecules, categories = job.results.get_network()
except FileError:
pass
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial imports
from typing import List
import scm.plams as plams
from scm.input_classes import engines
from scm.reactions_discovery import ReactionsDiscoveryJob
from rdkit import Chem
from rdkit.Chem import Draw
# Settings for displaying molecules in the notebook
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.ipython_useSVG = True
IPythonConsole.molSize = 250, 250
try:
from scm.plams import view # view molecule using AMSview in a Jupyter Notebook in AMS2026+
except ImportError:
from scm.plams import plot_molecule # plot molecule in a Jupyter Notebook in AMS2023+
def view(molecule, **kwargs):
plot_molecule(molecule)
# this line is not required in AMS2025+
plams.init()
# ## Helpers for showing molecules
def draw_molecules(molecules: List[plams.Molecule]):
smiles = [molecule.properties.smiles for molecule in molecules]
return draw_smiles(smiles)
def draw_smiles(smiles: List[str]):
rd_mols = [Chem.MolFromSmiles(s) for s in smiles]
return Draw.MolsToGridImage(rd_mols)
# ## The ReactionsDiscoveryJob class
job = ReactionsDiscoveryJob(name="MyDiscovery")
driver = job.input
md = driver.MolecularDynamics
# ## Setting up the reactants for molecular dynamics
md.NumSimulations = 4
build = md.BuildSystem
build.NumAtoms = 250
build.Density = 0.9
build.Molecule[0].SMILES = "O" # Water
build.Molecule[0].MoleFraction = 1
build.Molecule[1].SMILES = "NCCO" # MEA
build.Molecule[1].MoleFraction = 2
build.Molecule[2].SMILES = "O=C=O" # Carbondioxide
build.Molecule[2].MoleFraction = 3
draw_smiles([build.Molecule[i].SMILES.val for i in range(len(build.Molecule))])
# ## Setting up reactive molecular dynamics
md.Enabled = "Yes"
md.Type = "NanoReactor"
reactor = md.NanoReactor
reactor.NumCycles = 10
reactor.Temperature = 500
reactor.MinVolumeFraction = 0.6
# ## Setting up network extraction and ranking
network = driver.NetworkExtraction
network.Enabled = "Yes"
network.UseCharges = "Yes"
ranking = driver.ProductRanking
ranking.Enabled = "Yes"
# ## Selecting the AMS engine to use
engine = engines.ReaxFF()
engine.ForceField = "Glycine.ff"
engine.TaperBO = "Yes" # This is a really important setting for reaction analysis with ReaxFF potentials
driver.Engine = engine
# ## Running reactions discovery
result = job.run() # start the job
job.check() # check if job was succesful
# ## Obtain the results
graph, molecules, categories = result.get_network()
# ## Categories
#
# The categories are `Products` `Reactants` and `Unstable`, as described in the reactions discovery manual. `molecules` is a dictionary with keys equal to the categories and each concomitant value is a list of PLAMS molecules.
print(categories)
draw_molecules(molecules["Reactants"])
# ## Products
#
# These are the side products that reactions discovery found in the order as found by the ranking algorithm.
draw_molecules(molecules["Products"][:6])
# ## Unstable
#
# Unstable products were determined to not likely exist outside of reactive dynamics. This e.g. includes radicals or structures that don't form stable molecules in isolation. Not all unstable molecules have a sensible 2d structure, so instead we plot their 3d structure.
view(molecules["Unstable"][0], width=200, height=200, picture_path="picture1.png")
view(molecules["Unstable"][1], width=200, height=200, picture_path="picture2.png")
view(molecules["Unstable"][2], width=200, height=200, picture_path="picture3.png")
# ## Graph of the reaction network
#
# The graph is a bipartate networkx DiGraph with reaction and molecule nodes. This can be stored on disk in standard graph formats, e.g. `.gml`
import networkx as nx
nx.write_gml(graph, "reaction_network.gml")
# ## Load a job not originally run by PLAMS
from scm.plams import FileError
try:
job = ReactionsDiscoveryJob.load_external("plams_workdir/MyDiscovery")
graph, molecules, categories = job.results.get_network()
except FileError:
pass