Active Learning: Liquid Water Properties¶
Train an active-learning M3GNet model for liquid water and validate it against diffusion, radial distribution functions, and density from molecular-dynamics simulations.
Trained model: M3GNet, starting from the Universal Potential (UP)
Reference method: ReaxFF Water2017.ff; J. Phys. Chem. B, 2017, 121 (24), pp 6021–6032
System: Liquid water at T = 300 K
Property |
Reference |
Retrained M3GNet |
M3GNet-UP |
Experiment |
|---|---|---|---|---|
Self-diffusion (10⁻⁹ m² s⁻¹) |
2.6 |
2.5 |
0.23 |
2.3 |
Density (g cm⁻³) |
1.01 |
1.02 |
0.95 |
1.00 |
Expected duration: This notebook takes about 24 hours to run on the CPU. This includes both the active learning and the production simulations to get the diffusion coefficient, radial distribution functions, and density.
When running active learning it’s usually a good idea to start off “simple” and make the system/structures gradually more complicated.
To train liquid water, we here
first train a potential at slightly above room temperature and 1.0 g/cm^3
continue with a second active learning loop where the density is explicitly scanned from a low to a high value
Initial imports¶
import scm.plams as plams
from scm.simple_active_learning import SimpleActiveLearningJob
import matplotlib.pyplot as plt
plams.init()
PLAMS working folder: /path/to/plams_workdir.003
Create an initial water box¶
water = plams.from_smiles("O")
for at in water:
at.properties = plams.Settings()
plams.plot_molecule(water)

box = plams.packmol(water, n_molecules=48, density=1.0)
plams.plot_molecule(box, rotation="-5x,5y,0z")

Let’s run a short MD simulation with M3GNet-UP-2022 to make the structure more realistic:
up_s = plams.Settings()
up_s.input.MLPotential.Model = "M3GNet-UP-2022"
up_s.runscript.nproc = 1 # always run AMS Driver in serial for MLPotential
up_md = plams.AMSNVTJob(
molecule=box,
settings=up_s,
name="up_md",
nsteps=1000,
timestep=0.5,
temperature=350,
)
up_md.run();
[22.02|09:51:37] JOB up_md STARTED
[22.02|09:51:37] JOB up_md RUNNING
[22.02|09:52:19] JOB up_md FINISHED
[22.02|09:52:20] JOB up_md SUCCESSFUL
<scm.plams.interfaces.adfsuite.ams.AMSResults at 0x7fccbdae5a90>
New structure:
starting_structure = up_md.results.get_main_molecule()
plams.plot_molecule(starting_structure, rotation="-5x,5y,0z")
Simple Active Learning setup¶
Reference engine settings¶
Here, we choose to train against ReaxFF Water-2017.ff, which gives a good water structure.
fast_ref_s = plams.Settings()
fast_ref_s.input.ReaxFF.ForceField = "Water2017.ff"
fast_ref_s.runscript.nproc = 1
slow_ref_s = plams.Settings()
slow_ref_s.input.QuantumESPRESSO.Pseudopotentials.Family = "SSSP-Efficiency"
slow_ref_s.input.QuantumESPRESSO.Pseudopotentials.Functional = "PBE"
slow_ref_s.input.QuantumESPRESSO.K_Points._h = "gamma"
slow_ref_s.input.QuantumESPRESSO.System = plams.Settings(
input_dft="revpbe", ecutwfc=40, vdw_corr="Grimme-D3", dftd3_version=4
)
Change to slow_ref_s to train to revPBE-D3(BJ) instead:
ref_s = fast_ref_s.copy()
NVT molecular dynamics settings¶
nvt_md_s = plams.AMSNVTJob(
nsteps=20000,
timestep=0.5,
temperature=(270, 350, 350),
tau=100,
thermostat="Berendsen",
).settings
ParAMS machine learning settings¶
ml_s = plams.Settings()
ml_s.input.ams.MachineLearning.Backend = "M3GNet"
ml_s.input.ams.MachineLearning.CommitteeSize = 1
ml_s.input.ams.MachineLearning.M3GNet.Model = "UniversalPotential"
ml_s.input.ams.MachineLearning.MaxEpochs = 200
Active learning settings¶
Liquid water is a “simple” homogeneous system, so we can expect the ML method to perform quite well. We therefore decrease the success criteria thresholds a bit compared to the default vvalues, to ensure that we get accurate results.
Since we will immediately continue with another active learning loop, we disable the “RerunSimulation” as we are not interested in the MD simulation per se.
al_s = plams.Settings()
al_s.input.ams.ActiveLearning.Steps.Type = "Geometric"
al_s.input.ams.ActiveLearning.Steps.Geometric.Start = 10
al_s.input.ams.ActiveLearning.Steps.Geometric.NumSteps = 8
al_s.input.ams.ActiveLearning.InitialReferenceData.Generate.M3GNetShortMD.Enabled = (
"Yes"
)
al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.003
al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.35
al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
Complete job¶
settings = ref_s + nvt_md_s + ml_s + al_s
job = SimpleActiveLearningJob(
settings=settings, molecule=starting_structure, name="sal"
)
print(job.get_input())
ActiveLearning
AtEnd
RerunSimulation False
End
InitialReferenceData
... output trimmed ....
0.0000000000 11.2817834662 0.0000000000
0.0000000000 0.0000000000 11.2817834662
End
End
Run the simple active learning job¶
job.run(watch=True);
[22.02|10:12:07] JOB sal STARTED
[22.02|10:12:07] Renaming job sal to sal.002
[22.02|10:12:07] JOB sal.002 RUNNING
[22.02|10:12:09] Simple Active Learning 2024.101, Nodes: 1, Procs: 1
[22.02|10:12:11] Composition of main system: H96O48
... output trimmed ....
[22.02|10:36:15] Active learning finished!
[22.02|10:36:15] Goodbye!
[22.02|10:36:15] JOB sal.002 FINISHED
[22.02|10:36:15] JOB sal.002 SUCCESSFUL
<scm.simple_active_learning.plams.simple_active_learning_job.SimpleActiveLearningResults at 0x7fccbe25c580>
Validate trained model by RDF and MSD¶
Note: You should skip this part if you trained to DFT since the reference MD calculation will take a very long time!
mol = job.results.get_main_molecule()
plams.plot_molecule(mol, rotation="-5x,5y,0z")
retrained_model_settings = (
job.results.get_params_job().results.get_production_engine_settings()
)
retrained_model_settings.runscript.nproc = 1
Equilibration and production MD settings¶
eq_md_settings = plams.AMSNVTJob(
nsteps=8000,
timestep=0.5,
thermostat="Berendsen",
tau=100,
temperature=300,
samplingfreq=100,
).settings
prod_md_settings = plams.AMSNVTJob(
nsteps=50000,
timestep=0.5,
thermostat="NHC",
tau=200,
temperature=300,
samplingfreq=100,
).settings
Retrained model equilibration¶
retrained_model_eq_md_job = plams.AMSJob(
settings=eq_md_settings + retrained_model_settings,
molecule=mol,
name="retrained_model_eq_md_dens_1",
)
retrained_model_eq_md_job.run();
[22.02|13:39:24] JOB eq_md_dens_1 STARTED
[22.02|13:39:24] Renaming job eq_md_dens_1 to eq_md_dens_1.003
[22.02|13:39:24] JOB eq_md_dens_1.003 RUNNING
[22.02|13:42:58] JOB eq_md_dens_1.003 FINISHED
[22.02|13:42:58] JOB eq_md_dens_1.003 SUCCESSFUL
Retrained model production simulation¶
Let’s then run a production simulation from the final structure of the above equilibration MD using both the retrained model and the reference engine:
retrained_model_prod_md_job = plams.AMSJob(
settings=prod_md_settings + retrained_model_settings,
name="retrained_model_prod_md_dens_1",
molecule=retrained_model_eq_md_job.results.get_main_molecule(),
)
retrained_model_prod_md_job.run();
[22.02|13:46:22] JOB retrained_model_prod_md_dens_1 STARTED
[22.02|13:46:22] Renaming job retrained_model_prod_md_dens_1 to retrained_model_prod_md_dens_1.005
[22.02|13:46:22] JOB retrained_model_prod_md_dens_1.005 RUNNING
[22.02|14:07:30] JOB retrained_model_prod_md_dens_1.005 FINISHED
[22.02|14:07:30] JOB retrained_model_prod_md_dens_1.005 SUCCESSFUL
Reference equilibration MD¶
reference_eq_md_job = plams.AMSJob(
settings=eq_md_settings + ref_s,
molecule=mol,
name="reference_eq_md_dens_1",
)
reference_eq_md_job.run();
[22.02|14:08:55] JOB reference_eq_md_dens_1 STARTED
[22.02|14:08:55] JOB reference_eq_md_dens_1 RUNNING
[22.02|14:09:19] JOB reference_eq_md_dens_1 FINISHED
[22.02|14:09:19] JOB reference_eq_md_dens_1 SUCCESSFUL
Reference production MD¶
reference_prod_md_job = plams.AMSJob(
settings=prod_md_settings + ref_s,
name="reference_prod_md_dens_1",
molecule=reference_eq_md_job.results.get_main_molecule(),
)
reference_prod_md_job.run();
[22.02|14:09:19] JOB reference_prod_md_dens_1 STARTED
[22.02|14:09:19] Renaming job reference_prod_md_dens_1 to reference_prod_md_dens_1.003
[22.02|14:09:19] JOB reference_prod_md_dens_1.003 RUNNING
[22.02|14:11:59] JOB reference_prod_md_dens_1.003 FINISHED
[22.02|14:11:59] JOB reference_prod_md_dens_1.003 SUCCESSFUL
Mean squared displacement (MSD) helper functions¶
For a detailed explanation of the MSD and RDF jobs, see the “Molecular dynamics with Python” tutorial
def get_msd_job(job: plams.AMSJob, symbol: str = "O"):
atom_indices = [
i
for i, at in enumerate(job.results.get_main_molecule(), 1)
if at.symbol == symbol
]
msd_job = plams.AMSMSDJob(
job,
name="msd-" + job.name,
atom_indices=atom_indices, # indices start with 1 for the first atom
max_correlation_time_fs=4000, # max correlation time must be set before running the job
start_time_fit_fs=2000, # start_time_fit can also be changed later in the postanalysis
)
msd_job.run()
return msd_job
def plot_msd(job, start_time_fit_fs=None):
"""job: an AMSMSDJob"""
time, msd = job.results.get_msd()
fit_result, fit_x, fit_y = job.results.get_linear_fit(
start_time_fit_fs=start_time_fit_fs
)
# the diffusion coefficient can also be calculated as fit_result.slope/6 (ang^2/fs)
diffusion_coefficient = job.results.get_diffusion_coefficient(
start_time_fit_fs=start_time_fit_fs
) # m^2/s
plt.figure(figsize=(5, 3))
plt.plot(time, msd, label="MSD")
plt.plot(
fit_x, fit_y, label="Linear fit slope={:.5f} ang^2/fs".format(fit_result.slope)
)
plt.legend()
plt.xlabel("Correlation time (fs)")
plt.ylabel("Mean square displacement (ang^2)")
plt.title("MSD: Diffusion coefficient = {:.2e} m^2/s".format(diffusion_coefficient))
Temporarily turn off PLAMS logging¶
Technically, the MSD and RDF jobs are normal PLAMS jobs. However, they are very fast to run. We can turn off the PLAMS logging to keep the Jupyter notebook a bit more tidy:
plams.config.log.stdout = 0
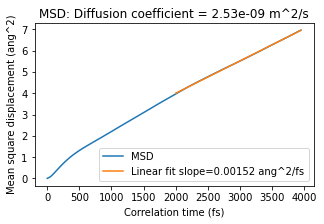
Retrained model MSD, diffusion coefficient¶
retrained_model_msd_job = get_msd_job(retrained_model_prod_md_job, "O")
retrained_model_D = (
retrained_model_msd_job.results.get_diffusion_coefficient()
) # diffusion coefficient, m^2/s
plot_msd(retrained_model_msd_job)
Reference MSD, diffusion coefficient¶
reference_msd_job = get_msd_job(reference_prod_md_job, "O")
reference_D = (
reference_msd_job.results.get_diffusion_coefficient()
) # diffusion coefficient, m^2/s
plot_msd(reference_msd_job)
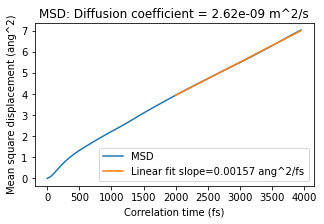
Conclusion for diffusion coefficient: In this case, the retrained model gives 2.53e-9 m^2/s and the reference method 2.62e-9 m^2/s. That is very good agreement! Note: your results may be somewhat different.
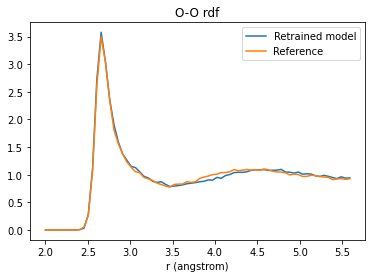
Retrained model and reference RDF¶
Let’s compare the calculated O-O, O-H, and H-H radial distribution functions (rdf):
def get_rdf(job, atom_indices, atom_indices_to, rmin, rmax, rstep):
rdf = plams.AMSRDFJob(
job,
atom_indices=atom_indices,
atom_indices_to=atom_indices_to,
rmin=rmin,
rmax=rmax,
rstep=rstep,
)
rdf.run()
return rdf.results.get_rdf()
final_frame = (
retrained_model_prod_md_job.results.get_main_molecule()
) # doesn't matter if retrained model or reference
O_ind = [i for i, at in enumerate(final_frame, 1) if at.symbol == "O"]
H_ind = [i for i, at in enumerate(final_frame, 1) if at.symbol == "H"]
rmax = final_frame.lattice[0][0] / 2
rstep = 0.05
O-O rdf¶
atom_indices, atom_indices_to = O_ind, O_ind
rmin = 2.0
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("O-O rdf");
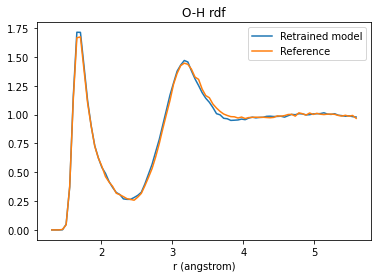
O-H rdf¶
atom_indices, atom_indices_to = O_ind, H_ind
rmin = 1.3
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("O-H rdf");
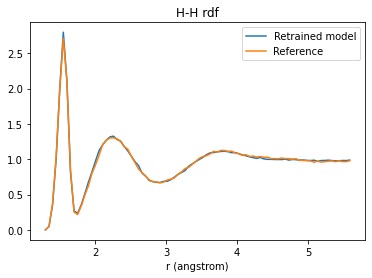
H-H rdf¶
atom_indices, atom_indices_to = H_ind, H_ind
rmin = 1.3
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("H-H rdf");
Turn PLAMS logging back on¶
plams.config.log.stdout = 3 # default value
Density and NPT¶
Check the predicted vs. reference density¶
npt_md_s = plams.AMSNPTJob(
nsteps=100000,
timestep=0.5,
thermostat="NHC",
tau=100,
temperature=300,
barostat="MTK",
barostat_tau=1000,
equal="XYZ",
pressure=1e5,
).settings
retrained_model_npt_job = plams.AMSJob(
settings=npt_md_s + retrained_model_settings,
name="retrained_model_npt",
molecule=retrained_model_prod_md_job.results.get_main_molecule(),
)
retrained_model_npt_job.run();
[22.02|15:25:22] JOB retrained_model_npt STARTED
[22.02|15:25:22] JOB retrained_model_npt RUNNING
[22.02|16:06:01] JOB retrained_model_npt FINISHED
[22.02|16:06:01] JOB retrained_model_npt SUCCESSFUL
<scm.plams.interfaces.adfsuite.ams.AMSResults at 0x7fcc8c910670>
retrained_model_density = (
plams.AMSNPTJob.load_external(retrained_model_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(
f"Retrained model water density at 300 K: {retrained_model_density * 1e-3:.2f} g/cm^3"
)
Retrained model water density at 300 K: 0.95 g/cm^3
plams.config.jobmanager.hashing = None
reference_npt_job = plams.AMSJob(
settings=npt_md_s + ref_s,
name="reference_npt",
molecule=reference_prod_md_job.results.get_main_molecule(),
)
reference_npt_job.run()
[22.02|18:53:54] JOB reference_npt STARTED
[22.02|18:53:54] Renaming job reference_npt to reference_npt.002
[22.02|18:53:54] JOB reference_npt.002 RUNNING
[22.02|18:58:13] JOB reference_npt.002 FINISHED
[22.02|18:58:13] JOB reference_npt.002 SUCCESSFUL
<scm.plams.interfaces.adfsuite.ams.AMSResults at 0x7fcc8d39d520>
reference_density = (
plams.AMSNPTJob.load_external(reference_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(f"Reference model water density at 300 K: {reference_density * 1e-3:.2f} g/cm^3")
Reference model water density at 300 K: 1.01 g/cm^3
The above reference value for ReaxFF Water-2017.ff agrees exactly with the published reference value of 1.01 g/cm^3.
However, the retrained M3GNet model predicts a density of 0.95 g/cm^3. The agreement is reasonable but not excellent. This can be explained by the fact that almost all training data points were at 1.00 g/cm^3. Only a few points (from the “M3GNetShortMD” initial reference data generator) were taken at other densities.
Let’s continue the active learning while sampling more densities. There are two strategies:
Use an NPT simulation during the active learning
Scan the density during the active learning
Here, we choose the second approach in order to ensure that multiple different densities are sampled.
Initial structure for scanning density¶
Get the final frame from one of the previous MD simulations, and linearly scale the density to 800 kg/m^3 = 0.8 g/cm^3. This will stretch out the O-H bonds so follow up with a short UFF preoptimization.
new_structure = final_frame.copy()
new_structure.set_density(850)
new_structure = plams.preoptimize(new_structure, model="uff", maxiterations=20)
plams.plot_molecule(new_structure)
Second active learning job: scanning density¶
Here we set Steps.Type = “Linear” to run reference calculations every 5000 MD steps.
To get an accurate density it’s very important that the predicted energy is accurate. It is not enough to just get a good fit for the forces.
Here, we decrease the success criteria for both the energy and forces compared to default values.
nsteps = 80000
scan_density_md_s = plams.AMSMDScanDensityJob(
molecule=new_structure,
scan_density_upper=1.15,
nsteps=nsteps,
tau=100,
thermostat="Berendsen",
temperature=300,
).settings
# we must explicitly set the StopStep, since the AL divides the simulation into multiple segments
scan_density_md_s.input.ams.MolecularDynamics.Deformation.StopStep = nsteps
# job = SimpleActiveLearningJob.load_external(plams.config.default_jobmanager.workdir + "/sal.002")
scan_density_ml_s = ml_s.copy()
scan_density_ml_s.input.ams.MachineLearning.LoadModel = (
job.results.get_params_results_directory()
)
scan_density_ml_s.input.ams.MachineLearning.Target.Forces.MAE = 0.02
scan_density_ml_s.input.ams.MachineLearning.MaxEpochs = 200
scan_density_al_s = plams.Settings()
scan_density_al_s.input.ams.ActiveLearning.Steps.Type = "Linear"
scan_density_al_s.input.ams.ActiveLearning.Steps.Linear.Start = 500
scan_density_al_s.input.ams.ActiveLearning.Steps.Linear.StepSize = 5000
scan_density_al_s.input.ams.ActiveLearning.InitialReferenceData.Load.FromPreviousModel = "Yes"
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.001
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Total = 0.002
# because we do not set Normalization, the above Energy criteria are energies per atom
# scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Normalization =
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.30
scan_density_al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
scan_density_al_s.input.ams.ActiveLearning.MaxReferenceCalculationsPerAttempt = 2
scan_density_al_job = SimpleActiveLearningJob(
name="scan_density_al",
settings=ref_s + scan_density_md_s + scan_density_ml_s + scan_density_al_s,
molecule=new_structure,
)
scan_density_al_job.run(watch=True);
[23.02|13:01:42] Simple Active Learning 2024.101, Nodes: 1, Procs: 1
[23.02|13:01:44] Composition of main system: H96O48
[23.02|13:01:44] All REFERENCE calculations will be performed with the following ReaxFF engine:
[23.02|13:01:44]
Engine reaxff
... output trimmed ....
[23.02|14:07:16] Active learning finished!
[23.02|14:07:16] Goodbye!
Let’s recalculate the density again:
new_retrained_model_settings = scan_density_al_job.results.get_params_job().results.get_production_engine_settings()
new_retrained_model_npt_job = plams.AMSJob(
settings=npt_md_s + new_retrained_model_settings,
name="new_retrained_model_npt",
molecule=retrained_model_prod_md_job.results.get_main_molecule(),
)
new_retrained_model_npt_job.run();
new_retrained_model_density = (
plams.AMSNPTJob.load_external(new_retrained_model_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(
f"New retrained model water density at 300 K: {new_retrained_model_density * 1e-3:.2f} g/cm^3"
)
New retrained model water density at 300 K: 1.02 g/cm^3
Conclusion for the density: Using active learning when scanning the densities makes sure that the predicitons are accurate for all densities. Consequently the equilibrium density is also in better agreement with the reference value of 1.01 g/cm^3.
Note that the density in general is quite difficult to fit accurately.
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# When running active learning it's usually a good idea to start off "simple" and make the system/structures gradually more complicated.
#
# To train liquid water, we here
#
# * first train a potential at slightly above room temperature and 1.0 g/cm^3
#
# * continue with a second active learning loop where the density is explicitly scanned from a low to a high value
#
# ## Initial imports
import scm.plams as plams
from scm.simple_active_learning import SimpleActiveLearningJob
import matplotlib.pyplot as plt
plams.init()
# ## Create an initial water box
water = plams.from_smiles("O")
for at in water:
at.properties = plams.Settings()
plams.plot_molecule(water)
box = plams.packmol(water, n_molecules=48, density=1.0)
plams.plot_molecule(box, rotation="-5x,5y,0z")
# Let's run a short MD simulation with M3GNet-UP-2022 to make the structure more realistic:
up_s = plams.Settings()
up_s.input.MLPotential.Model = "M3GNet-UP-2022"
up_s.runscript.nproc = 1 # always run AMS Driver in serial for MLPotential
up_md = plams.AMSNVTJob(
molecule=box,
settings=up_s,
name="up_md",
nsteps=1000,
timestep=0.5,
temperature=350,
)
up_md.run();
# New structure:
starting_structure = up_md.results.get_main_molecule()
plams.plot_molecule(starting_structure, rotation="-5x,5y,0z")
# ## Simple Active Learning setup
#
# ### Reference engine settings
#
# Here, we choose to train against ReaxFF Water-2017.ff, which gives a good water structure.
fast_ref_s = plams.Settings()
fast_ref_s.input.ReaxFF.ForceField = "Water2017.ff"
fast_ref_s.runscript.nproc = 1
slow_ref_s = plams.Settings()
slow_ref_s.input.QuantumESPRESSO.Pseudopotentials.Family = "SSSP-Efficiency"
slow_ref_s.input.QuantumESPRESSO.Pseudopotentials.Functional = "PBE"
slow_ref_s.input.QuantumESPRESSO.K_Points._h = "gamma"
slow_ref_s.input.QuantumESPRESSO.System = plams.Settings(
input_dft="revpbe", ecutwfc=40, vdw_corr="Grimme-D3", dftd3_version=4
)
# Change to slow_ref_s to train to revPBE-D3(BJ) instead:
ref_s = fast_ref_s.copy()
# ### NVT molecular dynamics settings
nvt_md_s = plams.AMSNVTJob(
nsteps=20000,
timestep=0.5,
temperature=(270, 350, 350),
tau=100,
thermostat="Berendsen",
).settings
# ### ParAMS machine learning settings
ml_s = plams.Settings()
ml_s.input.ams.MachineLearning.Backend = "M3GNet"
ml_s.input.ams.MachineLearning.CommitteeSize = 1
ml_s.input.ams.MachineLearning.M3GNet.Model = "UniversalPotential"
ml_s.input.ams.MachineLearning.MaxEpochs = 200
# ### Active learning settings
#
# Liquid water is a "simple" homogeneous system, so we can expect the ML method to perform quite well. We therefore decrease the success criteria thresholds a bit compared to the default vvalues, to ensure that we get accurate results.
#
# Since we will immediately continue with another active learning loop, we disable the "RerunSimulation" as we are not interested in the MD simulation per se.
al_s = plams.Settings()
al_s.input.ams.ActiveLearning.Steps.Type = "Geometric"
al_s.input.ams.ActiveLearning.Steps.Geometric.Start = 10
al_s.input.ams.ActiveLearning.Steps.Geometric.NumSteps = 8
al_s.input.ams.ActiveLearning.InitialReferenceData.Generate.M3GNetShortMD.Enabled = (
"Yes"
)
al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.003
al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.35
al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
# ### Complete job
settings = ref_s + nvt_md_s + ml_s + al_s
job = SimpleActiveLearningJob(
settings=settings, molecule=starting_structure, name="sal"
)
print(job.get_input())
# ### Run the simple active learning job
job.run(watch=True);
# ## Validate trained model by RDF and MSD
#
# Note: You should skip this part if you trained to DFT since the reference MD calculation will take a very long time!
mol = job.results.get_main_molecule()
plams.plot_molecule(mol, rotation="-5x,5y,0z")
retrained_model_settings = (
job.results.get_params_job().results.get_production_engine_settings()
)
retrained_model_settings.runscript.nproc = 1
# ### Equilibration and production MD settings
eq_md_settings = plams.AMSNVTJob(
nsteps=8000,
timestep=0.5,
thermostat="Berendsen",
tau=100,
temperature=300,
samplingfreq=100,
).settings
prod_md_settings = plams.AMSNVTJob(
nsteps=50000,
timestep=0.5,
thermostat="NHC",
tau=200,
temperature=300,
samplingfreq=100,
).settings
# ### Retrained model equilibration
retrained_model_eq_md_job = plams.AMSJob(
settings=eq_md_settings + retrained_model_settings,
molecule=mol,
name="retrained_model_eq_md_dens_1",
)
retrained_model_eq_md_job.run();
# ### Retrained model production simulation
# Let's then run a production simulation from the final structure of the above equilibration MD using both the retrained model and the reference engine:
retrained_model_prod_md_job = plams.AMSJob(
settings=prod_md_settings + retrained_model_settings,
name="retrained_model_prod_md_dens_1",
molecule=retrained_model_eq_md_job.results.get_main_molecule(),
)
retrained_model_prod_md_job.run();
# ### Reference equilibration MD
reference_eq_md_job = plams.AMSJob(
settings=eq_md_settings + ref_s,
molecule=mol,
name="reference_eq_md_dens_1",
)
reference_eq_md_job.run();
# ### Reference production MD
reference_prod_md_job = plams.AMSJob(
settings=prod_md_settings + ref_s,
name="reference_prod_md_dens_1",
molecule=reference_eq_md_job.results.get_main_molecule(),
)
reference_prod_md_job.run();
# ### Mean squared displacement (MSD) helper functions
# For a detailed explanation of the MSD and RDF jobs, see the "Molecular dynamics with Python" tutorial
def get_msd_job(job: plams.AMSJob, symbol: str = "O"):
atom_indices = [
i
for i, at in enumerate(job.results.get_main_molecule(), 1)
if at.symbol == symbol
]
msd_job = plams.AMSMSDJob(
job,
name="msd-" + job.name,
atom_indices=atom_indices, # indices start with 1 for the first atom
max_correlation_time_fs=4000, # max correlation time must be set before running the job
start_time_fit_fs=2000, # start_time_fit can also be changed later in the postanalysis
)
msd_job.run()
return msd_job
def plot_msd(job, start_time_fit_fs=None):
"""job: an AMSMSDJob"""
time, msd = job.results.get_msd()
fit_result, fit_x, fit_y = job.results.get_linear_fit(
start_time_fit_fs=start_time_fit_fs
)
# the diffusion coefficient can also be calculated as fit_result.slope/6 (ang^2/fs)
diffusion_coefficient = job.results.get_diffusion_coefficient(
start_time_fit_fs=start_time_fit_fs
) # m^2/s
plt.figure(figsize=(5, 3))
plt.plot(time, msd, label="MSD")
plt.plot(
fit_x, fit_y, label="Linear fit slope={:.5f} ang^2/fs".format(fit_result.slope)
)
plt.legend()
plt.xlabel("Correlation time (fs)")
plt.ylabel("Mean square displacement (ang^2)")
plt.title("MSD: Diffusion coefficient = {:.2e} m^2/s".format(diffusion_coefficient))
plt.gcf().savefig("picture1.png")
plt.clf()
# ### Temporarily turn off PLAMS logging
#
# Technically, the MSD and RDF jobs are normal PLAMS jobs. However, they are very fast to run. We can turn off the PLAMS logging to keep the Jupyter notebook a bit more tidy:
plams.config.log.stdout = 0
# ### Retrained model MSD, diffusion coefficient
retrained_model_msd_job = get_msd_job(retrained_model_prod_md_job, "O")
retrained_model_D = (
retrained_model_msd_job.results.get_diffusion_coefficient()
) # diffusion coefficient, m^2/s
plot_msd(retrained_model_msd_job)
# ### Reference MSD, diffusion coefficient
reference_msd_job = get_msd_job(reference_prod_md_job, "O")
reference_D = (
reference_msd_job.results.get_diffusion_coefficient()
) # diffusion coefficient, m^2/s
plot_msd(reference_msd_job)
# **Conclusion for diffusion coefficient**: In this case, the retrained model gives 2.53e-9 m^2/s and the reference method 2.62e-9 m^2/s. That is very good agreement! Note: your results may be somewhat different.
# ### Retrained model and reference RDF
#
# Let's compare the calculated O-O, O-H, and H-H radial distribution functions (rdf):
def get_rdf(job, atom_indices, atom_indices_to, rmin, rmax, rstep):
rdf = plams.AMSRDFJob(
job,
atom_indices=atom_indices,
atom_indices_to=atom_indices_to,
rmin=rmin,
rmax=rmax,
rstep=rstep,
)
rdf.run()
return rdf.results.get_rdf()
final_frame = (
retrained_model_prod_md_job.results.get_main_molecule()
) # doesn't matter if retrained model or reference
O_ind = [i for i, at in enumerate(final_frame, 1) if at.symbol == "O"]
H_ind = [i for i, at in enumerate(final_frame, 1) if at.symbol == "H"]
rmax = final_frame.lattice[0][0] / 2
rstep = 0.05
# ### O-O rdf
atom_indices, atom_indices_to = O_ind, O_ind
rmin = 2.0
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("O-O rdf");
plt.gcf().savefig("picture2.png")
plt.clf()
# ### O-H rdf
atom_indices, atom_indices_to = O_ind, H_ind
rmin = 1.3
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("O-H rdf");
plt.gcf().savefig("picture3.png")
plt.clf()
# ### H-H rdf
atom_indices, atom_indices_to = H_ind, H_ind
rmin = 1.3
pred_x, pred_y = get_rdf(
retrained_model_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
ref_x, ref_y = get_rdf(
reference_prod_md_job, atom_indices, atom_indices_to, rmin, rmax, rstep
)
plt.plot(pred_x, pred_y, label="Retrained model")
plt.plot(ref_x, ref_y, label="Reference")
plt.xlabel("r (angstrom)")
plt.legend()
plt.title("H-H rdf");
plt.gcf().savefig("picture4.png")
plt.clf()
# ### Turn PLAMS logging back on
plams.config.log.stdout = 3 # default value
# ## Density and NPT
#
# ### Check the predicted vs. reference density
npt_md_s = plams.AMSNPTJob(
nsteps=100000,
timestep=0.5,
thermostat="NHC",
tau=100,
temperature=300,
barostat="MTK",
barostat_tau=1000,
equal="XYZ",
pressure=1e5,
).settings
retrained_model_npt_job = plams.AMSJob(
settings=npt_md_s + retrained_model_settings,
name="retrained_model_npt",
molecule=retrained_model_prod_md_job.results.get_main_molecule(),
)
retrained_model_npt_job.run();
retrained_model_density = (
plams.AMSNPTJob.load_external(retrained_model_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(
f"Retrained model water density at 300 K: {retrained_model_density * 1e-3:.2f} g/cm^3"
)
plams.config.jobmanager.hashing = None
reference_npt_job = plams.AMSJob(
settings=npt_md_s + ref_s,
name="reference_npt",
molecule=reference_prod_md_job.results.get_main_molecule(),
)
reference_npt_job.run()
reference_density = (
plams.AMSNPTJob.load_external(reference_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(f"Reference model water density at 300 K: {reference_density * 1e-3:.2f} g/cm^3")
# The above reference value for ReaxFF Water-2017.ff agrees exactly with the published reference value of 1.01 g/cm^3.
#
# However, the retrained M3GNet model predicts a density of 0.95 g/cm^3. The agreement is reasonable but not excellent. This can be explained by the fact that almost all training data points were at 1.00 g/cm^3. Only a few points (from the "M3GNetShortMD" initial reference data generator) were taken at other densities.
#
# Let's continue the active learning while sampling more densities. There are two strategies:
#
# * Use an NPT simulation during the active learning
# * Scan the density during the active learning
#
# Here, we choose the second approach in order to ensure that multiple different densities are sampled.
# ### Initial structure for scanning density
#
# Get the final frame from one of the previous MD simulations, and linearly scale the density to 800 kg/m^3 = 0.8 g/cm^3. This will stretch out the O-H bonds so follow up with a short UFF preoptimization.
new_structure = final_frame.copy()
new_structure.set_density(850)
new_structure = plams.preoptimize(new_structure, model="uff", maxiterations=20)
plams.plot_molecule(new_structure)
# ### Second active learning job: scanning density
#
# Here we set Steps.Type = "Linear" to run reference calculations every 5000 MD steps.
#
# To get an accurate density it's very important that the predicted energy is accurate. It is not enough to just get a good fit for the forces.
#
# Here, we decrease the success criteria for both the energy and forces compared to default values.
nsteps = 80000
scan_density_md_s = plams.AMSMDScanDensityJob(
molecule=new_structure,
scan_density_upper=1.15,
nsteps=nsteps,
tau=100,
thermostat="Berendsen",
temperature=300,
).settings
# we must explicitly set the StopStep, since the AL divides the simulation into multiple segments
scan_density_md_s.input.ams.MolecularDynamics.Deformation.StopStep = nsteps
# job = SimpleActiveLearningJob.load_external(plams.config.default_jobmanager.workdir + "/sal.002")
scan_density_ml_s = ml_s.copy()
scan_density_ml_s.input.ams.MachineLearning.LoadModel = (
job.results.get_params_results_directory()
)
scan_density_ml_s.input.ams.MachineLearning.Target.Forces.MAE = 0.02
scan_density_ml_s.input.ams.MachineLearning.MaxEpochs = 200
scan_density_al_s = plams.Settings()
scan_density_al_s.input.ams.ActiveLearning.Steps.Type = "Linear"
scan_density_al_s.input.ams.ActiveLearning.Steps.Linear.Start = 500
scan_density_al_s.input.ams.ActiveLearning.Steps.Linear.StepSize = 5000
scan_density_al_s.input.ams.ActiveLearning.InitialReferenceData.Load.FromPreviousModel = "Yes"
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.001
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Total = 0.002
# because we do not set Normalization, the above Energy criteria are energies per atom
# scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Normalization =
scan_density_al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.30
scan_density_al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
scan_density_al_s.input.ams.ActiveLearning.MaxReferenceCalculationsPerAttempt = 2
scan_density_al_job = SimpleActiveLearningJob(
name="scan_density_al",
settings=ref_s + scan_density_md_s + scan_density_ml_s + scan_density_al_s,
molecule=new_structure,
)
scan_density_al_job.run(watch=True);
# Let's recalculate the density again:
new_retrained_model_settings = scan_density_al_job.results.get_params_job().results.get_production_engine_settings()
new_retrained_model_npt_job = plams.AMSJob(
settings=npt_md_s + new_retrained_model_settings,
name="new_retrained_model_npt",
molecule=retrained_model_prod_md_job.results.get_main_molecule(),
)
new_retrained_model_npt_job.run();
new_retrained_model_density = (
plams.AMSNPTJob.load_external(new_retrained_model_npt_job.results.rkfpath())
.results.get_equilibrated_molecule()
.get_density()
)
print(
f"New retrained model water density at 300 K: {new_retrained_model_density * 1e-3:.2f} g/cm^3"
)
# **Conclusion for the density**: Using active learning when scanning the densities makes sure that the predicitons are accurate for all densities. Consequently the equilibrium density is also in better agreement with the reference value of 1.01 g/cm^3.
#
# Note that the density in general is quite difficult to fit accurately.