VASP geometry optimization with PLAMS¶
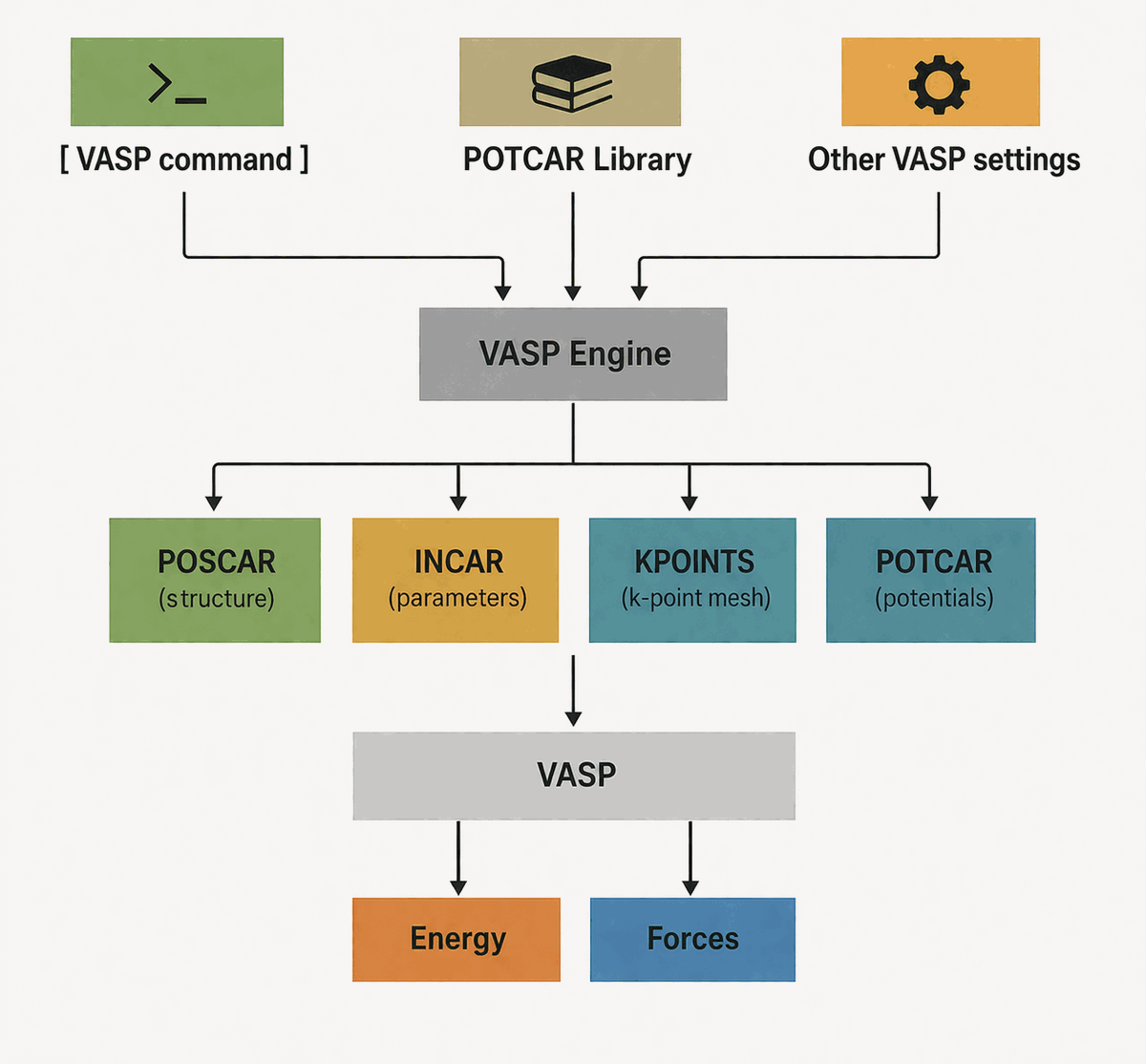
Set up a periodic geometry optimization with the VASP engine via PLAMS, assign
vasp.label values on a ChemicalSystem, inspect the generated INCAR,
KPOINTS, POTCAR, and POSCAR files, and see how the AMS driver
translates Engine VASP settings into the files used in the worker directory.
Set up imports and helper functions¶
This example runs a VASP geometry optimization through AMS and PLAMS and then inspects the generated VASP input files.
For a real VASP calculation, you must replace the VASPExec and POTCARLibrary values below with paths for your own VASP installation and POTCAR library. In this notebook they are intentionally set to bundled dummy example values so that you can focus on the workflow and inspect the generated VASP files.
import os
from pathlib import Path
from typing import Optional
import numpy as np
from scm.base import ChemicalSystem, Lattice
from scm.plams import AMSJob, Settings, view
AMSHOME = Path(os.environ["AMSHOME"])
AMSBIN = Path(os.environ["AMSBIN"])
def print_tree(root: Path, max_depth: Optional[int] = None, depth: int = 0) -> None:
"""Print a small directory tree rooted at ``root``."""
if depth == 0:
print(root)
if max_depth is not None and depth >= max_depth:
return
entries = sorted(root.iterdir(), key=lambda p: (p.is_file(), p.name.lower()))
for entry in entries:
indent = " " * depth
suffix = "/" if entry.is_dir() else ""
print(f"{indent}- {entry.name}{suffix}")
if entry.is_dir():
print_tree(entry, max_depth=max_depth, depth=depth + 1)
Build the starting ChemicalSystem¶
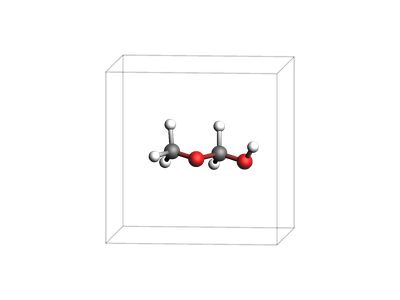
We start from ChemicalSystem.from_smiles("COCO"), add a cubic Lattice, and shift the atoms into the middle of the unit cell. We explicitly enable the vasp atom attributes on the ChemicalSystem and set atom.vasp.label = "O_h" for every oxygen atom - this will use the O_h (hard) POTCAR files from the POTCAR library.
system = ChemicalSystem.from_smiles("COCO")
system.lattice = Lattice([[8.0, 0.0, 0.0], [0.0, 8.0, 0.0], [0.0, 0.0, 8.0]])
system.perturb_coordinates(0.1)
system.translate([4.0, 4.0, 4.0])
system.bonds.clear_bonds()
system.enable_atom_attributes("vasp")
for atom in system:
if atom.symbol == "O":
atom.vasp.label = "O_h"
print(system)
System
Atoms
C 2.6856794203061445 3.9199419901846846 4.058575669511302
O 3.7601207518845516 3.5083608227031062 4.6401629714014945 vasp.label=O_h
C 4.933021513344075 3.872375390301085 3.7167210558876325
O 6.059029715136155 3.4526577254417194 4.254444615424399 vasp.label=O_h
H 2.45775147329223 3.5034676502464945 3.0599538733326
H 2.6106929948919837 5.173283492865718 3.98527725134316
H 1.7796445906520648 3.583834088594838 4.8894057730456595
H 4.941336171978787 5.159401738868146 3.5394929899243293
H 4.785515193212129 3.479040834222635 2.6738949779003613
H 6.368007465768469 4.055758604343035 5.05540326186522
End
Lattice
8 0 0
0 8 0
0 0 8
End
End
view(system, width=400, height=300, guess_bonds=True, direction="tilt_z")
Show the dummy VASPExec and POTCAR library used in this example¶
The current POTCARLibrary path is only meant as an example value. For your own calculations, point POTCARLibrary to your real VASP pseudopotential library, and point VASPExec to the command that launches your VASP executable.
VASP_EXEC_DUMMY = (
f"{AMSBIN}/amspython {AMSHOME}/scripting/scm/external_engines/backends/_vasp/fakevasp.py 6.4.0"
)
POTCAR_LIBRARY_DUMMY = AMSHOME / "atomicdata" / "VASP" / "test_potcars"
Command to execute VASP¶
print("Dummy VASPExec:")
print(VASP_EXEC_DUMMY)
print()
Dummy VASPExec:
/home/hellstrom/adfhome/bin/amspython /home/hellstrom/adfhome/scripting/scm/external_engines/backends/_vasp/fakevasp.py 6.4.0
POTCAR Library¶
print("Dummy POTCARLibrary:")
print(POTCAR_LIBRARY_DUMMY)
print()
print("Current POTCAR library structure:")
print_tree(POTCAR_LIBRARY_DUMMY, max_depth=2)
Dummy POTCARLibrary:
/home/hellstrom/adfhome/atomicdata/VASP/test_potcars
Current POTCAR library structure:
/home/hellstrom/adfhome/atomicdata/VASP/test_potcars
- C/
- POTCAR
- C_compressed/
- POTCAR.Z
- H/
- POTCAR
- H.75/
- POTCAR
- H1.25/
- POTCAR
- O/
- POTCAR
- O_h/
- POTCAR
- O_s/
- POTCAR
Build the PLAMS Settings object¶
The VASPExec and POTCARLibrary settings are essential for the VASP engine. In this notebook they use the dummy paths shown above, so replace them before using this workflow with a real VASP installation.
settings = Settings()
settings.input.ams.Task = "GeometryOptimization"
settings.input.VASP.EnergyCutoff = 400
settings.input.VASP.VASPExec = VASP_EXEC_DUMMY
settings.input.VASP.POTCARLibrary = str(POTCAR_LIBRARY_DUMMY)
settings.input.VASP.KPOINTSOrigin = "Gamma-centered"
settings.input.VASP.nk1 = 1
settings.input.VASP.nk2 = 1
settings.input.VASP.nk3 = 1
settings.input.VASP.Occupation = "Gaussian"
settings.input.VASP.Smearing = 0.01
job = AMSJob(settings=settings, molecule=system, name="vasp_geoopt")
print(job.get_input())
Task GeometryOptimization
System
Atoms
C 2.6856794203061445 3.9199419901846846 4.058575669511302
O 3.7601207518845516 3.5083608227031062 4.6401629714014945 vasp.label=O_h
C 4.933021513344075 3.872375390301085 3.7167210558876325
O 6.059029715136155 3.4526577254417194 4.254444615424399 vasp.label=O_h
H 2.45775147329223 3.5034676502464945 3.0599538733326
H 2.6106929948919837 5.173283492865718 3.98527725134316
H 1.7796445906520648 3.583834088594838 4.8894057730456595
H 4.941336171978787 5.159401738868146 3.5394929899243293
H 4.785515193212129 3.479040834222635 2.6738949779003613
H 6.368007465768469 4.055758604343035 5.05540326186522
End
Lattice
8 0 0
0 8 0
0 0 8
End
End
Engine VASP
EnergyCutoff 400
KPOINTSOrigin Gamma-centered
Occupation Gaussian
POTCARLibrary /home/hellstrom/adfhome/atomicdata/VASP/test_potcars
Smearing 0.01
VASPExec /home/hellstrom/adfhome/bin/amspython /home/hellstrom/adfhome/scripting/scm/external_engines/backends/_vasp/fakevasp.py 6.4.0
nk1 1
nk2 1
nk3 1
EndEngine
Run the geometry optimization¶
The AMS driver performs the geometry optimization, while the VASP engine writes the VASP-style files in the worker directory. In this documentation example, the engine command is the bundled fake-VASP executable.
results = job.run()
print(f"Job status ok: {results.ok()}")
print(f"Job directory: {job.path}")
[17.03|13:18:05] JOB vasp_geoopt STARTED
[17.03|13:18:05] JOB vasp_geoopt RUNNING
[17.03|13:18:10] JOB vasp_geoopt FINISHED
[17.03|13:18:10] JOB vasp_geoopt SUCCESSFUL
Job status ok: True
Job directory: /path/to/plams_workdir.004/vasp_geoopt
Inspect the job directory¶
The generated VASP files live inside the worker.0.0.0 subdirectory of the PLAMS job directory.
job_dir = Path(job.path)
worker_dir = next(job_dir.glob("worker.*"))
print_tree(job_dir, max_depth=2)
/path/to/plams_workdir.004/vasp_geoopt
- worker.0.0.0/
- def.py
- EngineVASP.log
- INCAR
- KPOINTS
- OUTCAR
- POSCAR
- POTCAR
- VASP.log
- ams.log
- ams.rkf
- input.json
- output.xyz
- system.system_in
- vasp.rkf
- vasp_geoopt.dill
- vasp_geoopt.err
- vasp_geoopt.in
- vasp_geoopt.out
- vasp_geoopt.run
Inspect INCAR, KPOINTS, and POTCAR¶
The files below are generated from the Engine VASP block and the atom labels on the ChemicalSystem:
INCARreflects keywords such asEnergyCutoff,Occupation, andSmearing.KPOINTSreflectsKPOINTSOrigin,nk1,nk2, andnk3.POTCARis assembled fromPOTCARLibrarytogether with the atom labels, so the oxygen atoms labeledO_hpick up theO_h/POTCARentry.
INCAR¶
print((worker_dir / "INCAR").read_text())
# INPUT GENERATED BY VASP INTERACTIVE
# MANDATORY INPUT
POTIM = 0.0
IBRION = -1
INTERACTIVE = .TRUE.
EDIFFG = 0
NSW = 999999999
# DEFAULT INPUT
IWAVPR = 11
ISYM = 0
ISIF = 3
# OPTIONAL INPUT
ISPIN = 1
ISMEAR = 0
ENCUT = 400.0
SIGMA = 0.01
PREC = Normal
IVDW = 0
KPOINTS¶
print((worker_dir / "KPOINTS").read_text())
# KPOINTS GENERATED BY VASP INTERACTIVE
0
Gamma
1 1 1
0.0 0.0 0.0
POTCAR¶
Note: each dummy POTCAR file is just a single line. Real POTCAR files are much bigger and have real data.
print((worker_dir / "POTCAR").read_text())
C POTCAR
O_h POTCAR
C POTCAR
O_h POTCAR
H POTCAR
Avoid looking at POSCAR¶
When running VASP via AMS, the POSCAR in the worker directory does not necessarily correspond to the original input structure. This is because sometimes VASP needs to be restarted (in case of changing composition, lattice, …).
Here is the POSCAR file for reference:
POSCAR¶
print((worker_dir / "POSCAR").read_text())
System POTCARS: C, O_h, C, O_h, H
1.0000000000000000
8.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 8.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 8.0000000000000000
C O C O H
1 1 1 1 6
Cartesian
2.6856794203061445 3.9199419901846846 4.0585756695113018
3.7601207518845516 3.5083608227031062 4.6401629714014945
4.9330215133440749 3.8723753903010851 3.7167210558876325
6.0590297151361554 3.4526577254417194 4.2544446154243989
2.4577514732922299 3.5034676502464945 3.0599538733326002
2.6106929948919837 5.1732834928657176 3.9852772513431600
1.7796445906520648 3.5838340885948381 4.8894057730456595
4.9413361719787874 5.1594017388681461 3.5394929899243293
4.7855151932121291 3.4790408342226349 2.6738949779003613
6.3680074657684687 4.0557586043430351 5.0554032618652203
In general, prefer the normal AMS methods to get the input or output systems from a finished job:
Input system¶
input_system = results.get_input_system()
print(input_system)
System
Atoms
C 2.6856794203061445 3.9199419901846846 4.058575669511302
O 3.7601207518845516 3.5083608227031062 4.6401629714014945 vasp.label=O_h
C 4.933021513344075 3.872375390301085 3.7167210558876325
O 6.059029715136155 3.4526577254417194 4.254444615424399 vasp.label=O_h
H 2.45775147329223 3.5034676502464945 3.0599538733326
H 2.6106929948919837 5.173283492865718 3.98527725134316
H 1.7796445906520648 3.583834088594838 4.8894057730456595
H 4.941336171978787 5.159401738868146 3.5394929899243293
H 4.785515193212129 3.479040834222635 2.6738949779003613
H 6.368007465768469 4.055758604343035 5.05540326186522
End
Lattice
8 0 0
0 8 0
0 0 8
End
End
Output system¶
optimized_system = results.get_main_system()
print(optimized_system)
System
Atoms
C 2.6329504031813014 3.9918774506365327 4.083412503363929
O 3.8660880228152315 3.534843359870515 4.591979968873295 vasp.label=O_h
C 4.904959292879035 3.921219069595505 3.716720304465348
O 6.128394423475451 3.465882633039338 4.221381820199423 vasp.label=O_h
H 2.414581165435873 3.5421932329137946 3.0891805654541704
H 2.609218201907933 5.102721856605182 4.0214124839331085
H 1.8261189342303186 3.6779111031336296 4.777176727109537
H 4.945202980489115 5.030700143042123 3.603445015365042
H 4.7597209992206 3.4636746564444505 2.7124977605685223
H 6.293564866831727 3.9770988324903884 5.056125290303781
End
Lattice
8 0 0
0 8 0
0 0 8
End
End
Appendix: sort system by vasp.label¶
It is common in VASP to sort the input structure by element (or, more precisely, by elemental POTCAR file). This reduces the size of the overall POTCAR file. You can do it like this:
sorted_system = system.copy()
sorted_system.enable_atom_attributes("vasp")
sorted_system.sort_atoms(lambda A, B: (A.vasp.label or A.symbol) < (B.vasp.label or B.symbol))
print(sorted_system)
System
Atoms
C 2.6856794203061445 3.9199419901846846 4.058575669511302
C 4.933021513344075 3.872375390301085 3.7167210558876325
H 2.45775147329223 3.5034676502464945 3.0599538733326
H 2.6106929948919837 5.173283492865718 3.98527725134316
H 1.7796445906520648 3.583834088594838 4.8894057730456595
H 4.941336171978787 5.159401738868146 3.5394929899243293
H 4.785515193212129 3.479040834222635 2.6738949779003613
H 6.368007465768469 4.055758604343035 5.05540326186522
O 3.7601207518845516 3.5083608227031062 4.6401629714014945 vasp.label=O_h
O 6.059029715136155 3.4526577254417194 4.254444615424399 vasp.label=O_h
End
Lattice
8 0 0
0 8 0
0 0 8
End
End
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Set up imports and helper functions
#
# This example runs a VASP geometry optimization through AMS and PLAMS and then inspects the generated VASP input files.
#
# For a **real** VASP calculation, you must replace the `VASPExec` and `POTCARLibrary` values below with paths for your own VASP installation and POTCAR library. In this notebook they are intentionally set to bundled **dummy** example values so that you can focus on the workflow and inspect the generated VASP files.
#
import os
from pathlib import Path
from typing import Optional
import numpy as np
from scm.base import ChemicalSystem, Lattice
from scm.plams import AMSJob, Settings, view
AMSHOME = Path(os.environ["AMSHOME"])
AMSBIN = Path(os.environ["AMSBIN"])
def print_tree(root: Path, max_depth: Optional[int] = None, depth: int = 0) -> None:
"""Print a small directory tree rooted at ``root``."""
if depth == 0:
print(root)
if max_depth is not None and depth >= max_depth:
return
entries = sorted(root.iterdir(), key=lambda p: (p.is_file(), p.name.lower()))
for entry in entries:
indent = " " * depth
suffix = "/" if entry.is_dir() else ""
print(f"{indent}- {entry.name}{suffix}")
if entry.is_dir():
print_tree(entry, max_depth=max_depth, depth=depth + 1)
# ## Build the starting `ChemicalSystem`
#
# We start from `ChemicalSystem.from_smiles("COCO")`, add a cubic `Lattice`, and shift the atoms into the middle of the unit cell. We explicitly enable the `vasp` atom attributes on the `ChemicalSystem` and set `atom.vasp.label = "O_h"` for every oxygen atom - this will use the `O_h` (hard) POTCAR files from the POTCAR library.
#
system = ChemicalSystem.from_smiles("COCO")
system.lattice = Lattice([[8.0, 0.0, 0.0], [0.0, 8.0, 0.0], [0.0, 0.0, 8.0]])
system.perturb_coordinates(0.1)
system.translate([4.0, 4.0, 4.0])
system.bonds.clear_bonds()
system.enable_atom_attributes("vasp")
for atom in system:
if atom.symbol == "O":
atom.vasp.label = "O_h"
print(system)
view(system, width=400, height=300, guess_bonds=True, direction="tilt_z", picture_path="picture1.png")
# ## Show the dummy VASPExec and POTCAR library used in this example
#
# The current `POTCARLibrary` path is only meant as an example value. For your own calculations, point `POTCARLibrary` to your real VASP pseudopotential library, and point `VASPExec` to the command that launches your VASP executable.
#
VASP_EXEC_DUMMY = f"{AMSBIN}/amspython {AMSHOME}/scripting/scm/external_engines/backends/_vasp/fakevasp.py 6.4.0"
POTCAR_LIBRARY_DUMMY = AMSHOME / "atomicdata" / "VASP" / "test_potcars"
# ### Command to execute VASP
print("Dummy VASPExec:")
print(VASP_EXEC_DUMMY)
print()
# ### POTCAR Library
print("Dummy POTCARLibrary:")
print(POTCAR_LIBRARY_DUMMY)
print()
print("Current POTCAR library structure:")
print_tree(POTCAR_LIBRARY_DUMMY, max_depth=2)
# ## Build the PLAMS `Settings` object
#
# The `VASPExec` and `POTCARLibrary` settings are essential for the VASP engine. In this notebook they use the dummy paths shown above, so replace them before using this workflow with a real VASP installation.
#
settings = Settings()
settings.input.ams.Task = "GeometryOptimization"
settings.input.VASP.EnergyCutoff = 400
settings.input.VASP.VASPExec = VASP_EXEC_DUMMY
settings.input.VASP.POTCARLibrary = str(POTCAR_LIBRARY_DUMMY)
settings.input.VASP.KPOINTSOrigin = "Gamma-centered"
settings.input.VASP.nk1 = 1
settings.input.VASP.nk2 = 1
settings.input.VASP.nk3 = 1
settings.input.VASP.Occupation = "Gaussian"
settings.input.VASP.Smearing = 0.01
job = AMSJob(settings=settings, molecule=system, name="vasp_geoopt")
print(job.get_input())
# ## Run the geometry optimization
#
# The AMS driver performs the geometry optimization, while the VASP engine writes the VASP-style files in the worker directory. In this documentation example, the engine command is the bundled fake-VASP executable.
#
results = job.run()
print(f"Job status ok: {results.ok()}")
print(f"Job directory: {job.path}")
# ## Inspect the job directory
#
# The generated VASP files live inside the `worker.0.0.0` subdirectory of the PLAMS job directory.
#
job_dir = Path(job.path)
worker_dir = next(job_dir.glob("worker.*"))
print_tree(job_dir, max_depth=2)
# ## Inspect `INCAR`, `KPOINTS`, and `POTCAR`
#
# The files below are generated from the `Engine VASP` block and the atom labels on the `ChemicalSystem`:
#
# - `INCAR` reflects keywords such as `EnergyCutoff`, `Occupation`, and `Smearing`.
# - `KPOINTS` reflects `KPOINTSOrigin`, `nk1`, `nk2`, and `nk3`.
# - `POTCAR` is assembled from `POTCARLibrary` together with the atom labels, so the oxygen atoms labeled `O_h` pick up the `O_h/POTCAR` entry.
#
# ### INCAR
print((worker_dir / "INCAR").read_text())
# ### KPOINTS
print((worker_dir / "KPOINTS").read_text())
# ### POTCAR
#
# Note: each dummy POTCAR file is just a single line. Real POTCAR files are much bigger and have real data.
print((worker_dir / "POTCAR").read_text())
# ## Avoid looking at `POSCAR`
#
# When running VASP via AMS, the `POSCAR` in the worker directory does **not** necessarily correspond to the original input structure. This is because sometimes VASP needs to be restarted (in case of changing composition, lattice, ...).
#
# Here is the POSCAR file for reference:
#
# ### POSCAR
print((worker_dir / "POSCAR").read_text())
# In general, **prefer the normal AMS methods** to get the input or output systems from a finished job:
# ### Input system
input_system = results.get_input_system()
print(input_system)
# ### Output system
optimized_system = results.get_main_system()
print(optimized_system)
# ## Appendix: sort system by vasp.label
#
# It is common in VASP to sort the input structure by element (or, more precisely, by elemental POTCAR file). This reduces the size of the overall POTCAR file. You can do it like this:
sorted_system = system.copy()
sorted_system.enable_atom_attributes("vasp")
sorted_system.sort_atoms(lambda A, B: (A.vasp.label or A.symbol) < (B.vasp.label or B.symbol))
print(sorted_system)