Restarts¶
The main results of a BAND calculation are stored in the rkf file. If you save this file you can use it to restart your calculation. The input for the restart calculation is essentially the same, except for some extra keys, like Restart, Grid, and DensityPlot.
Plots of the density (and many other symmetric properties) can can be obtained with the key DensityPlot. Density and orbital plot restarts require the specification of the Grid key.
With the subkey SCF you can start the SCF procedure with the last solution from the restart file. This can be useful if the SCF did not converge or if you want to compute some post-SCF properties (e.g. the DOS or the band structure). Similarly, a geometry optimization can be restarted with the subkey GeometryOptimization You can use the geometry of a previous calculation.
Usually the input for a restarted job is the same as for the original calculation, with some extra options, described in this section.
Some examples are available in the $AMSHOME/examples/band directory and are discussed in the Examples section.
Restart key¶
Restart
File string
SCF Yes/No
DensityPlot Yes/No
OrbitalPlot Yes/No
NOCVdRhoPlot Yes/No
NOCVOrbitalPlot Yes/No
UseDensityMatrix Yes/No
BandStructure Yes/No
DOS Yes/No
End
Restart- Type:
Block
- Description:
Tells the program that it should restart with the restart file, and what to restart.
File- Type:
String
- Default value:
- GUI name:
Restart using
- Description:
Name of the restart file. The file should be a band.rkf file from a previous run.
SCF- Type:
Bool
- Default value:
No
- GUI name:
Restart: SCF
- Description:
Continue the SCF procedure using the orbital coefficients and occupations from the restart file.
DensityPlot- Type:
Bool
- Default value:
No
- Description:
Goes together with the DensityPlot block and Grid blocks
OrbitalPlot- Type:
Bool
- Default value:
No
- Description:
Goes together with the OrbitalPlot and Grid
NOCVdRhoPlot- Type:
Bool
- Default value:
No
- Description:
Goes together with the NOCVdRhoPlot and Grid blocks.
NOCVOrbitalPlot- Type:
Bool
- Default value:
No
- Description:
Goes together with the NOCVOrbitalPlot and Grid blocks.
UseDensityMatrix- Type:
Bool
- Default value:
No
- Description:
If set to True: For restarting the SCF the density matrix will be used. Requires you to set ‘Save DensityMatrix’ in the previous run.
BandStructure- Type:
Bool
- Default value:
No
- Description:
Calculate the band structure from a previous calculation. Does not work with model potentials and Hubbard.
DOS- Type:
Bool
- Default value:
No
- Description:
Calculate the DOS from a previous calculation. Does not work with model potentials and Hubbard.
Grid¶
The Grid block is used for restart options OrbitalPlot, DensityPlot, NOCVOrbitalPlot and NOCVdRhoPlot. There are two ways to define your grid. The most easy way is to use the Type key, which automatically generates a grid around the atoms in the unit cell:
Grid
Type [coarse | medium | fine]
End
Grid- Type:
Block
- Description:
Options for the regular grid used for plotting (e.g. density plot). Used ICW the restart option.
Type- Type:
Multiple Choice
- Default value:
coarse
- Options:
[coarse, medium, fine]
- Description:
The default regular grids.
One alternative is to specify everything by hand via the ‘UserDefined’ sub-block.
Grid
UserDefined header # Non-standard block. See details.
...
End
End
Grid- Type:
Block
- Description:
Options for the regular grid used for plotting (e.g. density plot). Used ICW the restart option.
UserDefined- Type:
Non-standard block
- Description:
One can define the regular grid specification in this block. See example. Default unit is Bohr
The following input would create a cube from (-1,-1,-1) to (1,1,1) bohr:
Grid
UserDefined
-1 -1 -1 ! Starting point
1 0 0 0.1 ! vec1 and dvec1
0 1 0 0.1 ! vec2 and dvec2
0 0 1 0.1 ! vec3 and dvec3
20 20 20 ! nr. of steps along three directions
End
End
Note
The grid is specified in bohr
One can also specify a text file from which the grid is imported:
Grid
FileName string
End
Grid- Type:
Block
- Description:
Options for the regular grid used for plotting (e.g. density plot). Used ICW the restart option.
FileName- Type:
String
- Default value:
- Description:
Read in the grid from a file. The file format of the grid is: three numbers per line (defining the x, y and z coordinates of the points).
Plots of the density, potential, and many more properties¶
DensityPlot # Non-standard block. See details.
...
End
DensityPlot- Type:
Non-standard block
- Description:
Plots of the density. Goes together with the Restart%DensityPlot and Grid keys.
The DensityPlot block goes together with the Restart%DensityPlot and Grid keys. Example input:
...
Restart
File my_file.rkf
DensityPlot
End
Grid
Type Coarse
End
DensityPlot
rho(fit)
vxc[rho]
End
...
After such a run you get a TAPE41 file that you should rename to my.t41, and view with AMSview.
The most common properties to plot are:
rho(fit)The fitted density.v(coulomb)The Coulomb potential.vxc[rho(fit)]the XC potential (using the fitted density)vxc[rho]XC potential of the exact densityrhoThe density|gradRho|The norm of the gradient of the densitytauThe symmetric kinetic energy densityLDOSThe local density of states. (See LDOS key)elf[rho]The electron localization functionXThe Electron energy density function from Ref [1] [2] . EquivalentlyX(fit)may be used as an approximation, employing the density fit.
Some more specialized options are:
rho(deformation/fit)the fitted deformation densityrho(atoms)The density of the startup atomsv(coulomb/atoms)The Coulomb potential of the start densitys[rho]Reduced density gradient. Common ingredient for XC functionalss[rho(fit)]Same as above, now for the fit densityalpha[rho]Ingredient for some meta-GGAs
In the BAND example directory there is the Frags_COCu example which shows how this can be used in combination with the Fragment key.
Orbital plots¶
OrbitalPlot # Non-standard block. See details.
...
End
OrbitalPlot- Type:
Non-standard block
- Description:
Goes together with the Restart%OrbitalPlot and Grid keys. See Example.
The OrbitalPlot block goes together with the Restart%OrbitalPlot and Grid keys. Example input:
...
Restart
File my_file.rkf
OrbitalPlot
End
Grid
Type Coarse
End
OrbitalPlot
1 Band 5 8 ! for k-point 1 plot bands 5 to 8
5 Band 6 ! for k-point 5 plot band 6
6 -0.2 +0.3 ! for k-point 6 plot bands between -0.2 and +0.3 a.u. w.r.t Fermi level
End
...
After such a run you get a TAPE41 file that you should rename to my.t41, and view with AMSview.
Induced Density Plots of Response Calculations¶
ResponseInducedDensityPlot # Non-standard block. See details.
...
End
ResponseInducedDensityPlot- Type:
Non-standard block
- Description:
Goes together with Restart%ResponseInducedDensityPlot and Grid.
ResponseInducedDensityPlot (block-type)
The ResponseInducedDensityPlot block goes together with the Restart%ResponseInducedDensityPlot and Grid keys. In the BAND example directory there is the TD-CDFT for MoS2 Monolayer example that shows how this can be used. Example input:
...
Restart
File my_file.rkf
ResponseInducedDensityPlot
End
Grid
Type Coarse
End
ResponseInducedDensityPlot
XCOMPONENT 5 8 ! plot x component of induced densities
! for frequencies number 5 to 8
YCOMPONENT 6 ! plot y component of induced densities
! for frequency number 6
ZCOMPONENT 1 ! plot z component of induced densities
! for frequency number 1
End
...
After such a run you get a TAPE41 file that you should rename to my.t41, and view with AMSview.
Attention
The plotting capability works only with response calculation RUNKF files based on the NewResponse method!
NOCV Orbital Plots¶
NOCVOrbitalPlot # Non-standard block. See details.
...
End
NOCVOrbitalPlot- Type:
Non-standard block
- Description:
Goes together with the Restart%NOCVOrbitalPlot and Grid keys. See example.
The NOCVOrbitalPlot blockg oes together with the Restart%NOCVOrbitalPlot and Grid keys. See example PEDANOCV_MgO+CO. Example input:
...
Restart
File my_file.rkf
NOCVOrbitalPlot
End
Grid
Type Coarse
End
NOCVOrbitalPlot
1 Band 5 8 ! for k-point 1 plot NOCV Orbitals 5 to 8
End
...
After such a run you get a TAPE41 file that you should rename to my.t41, and view with AMSview.
NOCV Deformation Density Plots¶
NOCVdRhoPlot # Non-standard block. See details.
...
End
NOCVdRhoPlot- Type:
Non-standard block
- Description:
Goes together with the Restart%NOCVdRhoPlot and Grid keys. See example.
The NOCVdRhoPlot blockg oes together with the Restart%NOCVdRhoPlot and Grid keys. See example PEDANOCV_MgO+CO. Example input:
...
Restart
File my_file.rkf
NOCVdRhoPlot
End
Grid
Type Coarse
End
NOCVdRhoPlot
1 Band 5 8 ! for k-point 1 plot NOCV deformation densities 5 to 8
End
...
After such a run you get a TAPE41 file that you should rename to my.t41, and view with AMSview.
LDOS (STM)¶
The local density of states (LDOS) represents a partial density, (see wikipedia): it is the density arising from states within an energy window.
LDOS
DeltaNeg float
DeltaPos float
Shift float
End
LDOS- Type:
Block
- Description:
Local Density-Of-States information. This can be used to generate STM images in the Tersoff-Hamann approximation (see https://doi.org/10.1103/PhysRevB.31.805)
DeltaNeg- Type:
Float
- Default value:
0.0001
- Unit:
Hartree
- Description:
Lower bound energy (Shift-DeltaNeg)
DeltaPos- Type:
Float
- Default value:
0.0001
- Unit:
Hartree
- Description:
Upper bound energy (Shift+DeltaPos)
Shift- Type:
Float
- Default value:
0.0
- Unit:
Hartree
- Description:
The energy bias with respect to the Fermi level.
Integrating from minus infinity (DeltaNeg=1e6) to the fermi level (DeltaPos=0) produces the total (valence) density.
The local density of states is integrated over the resulting interval. Example of an LDOS restart:
Restart
File my_file.rkf
DensityPlot
End
Grid
Type Coarse
End
DensityPlot
LDOS
End
LDOS
Shift 0.1
DeltaNeg 0.001
DeltaPos 0.0
End
According to this example, we restart from the result file of a previous calculation. The calculation will generate a file TAPE41 which can be viewed with AMSview. (Rename the file to my.t41)
See also Restart, and DensityPlot.
Electron Energy Density¶
The electron energy density is defined as [1]
\(X(r) = -\left\{ \frac{1}{2} \sum_i^\text{occ} \nabla \psi_i \cdot \nabla \psi_i -\frac{1}{4}\nabla^2\rho - V_\text{effective} \rho \right\}\)
It can be obtained by requesting X or X(fit) in a restart, see also Restart, and DensityPlot.
Save¶
Save string
Save- Type:
String
- Recurring:
True
- Description:
Save scratch files or extra data that would be otherwise deleted at the end of the calculation. e.g. ‘TAPE10’ (containing the integration grid) or ‘DensityMatrix’
Restarting the DOS and/or BandStructure¶
Perhaps you did a calculation, optimizing the geometry, and now want to see the band structure and partial DOS. This can be achieved by using Restart%DOS and Restart%BandStructure. This way you can easily refine your plots, or solve a missing DOS problem, without having to repeat the whole SCF.
With the restarting of the DOS there is the special possibility to use a better k-grid than was used during the SCF. Whereas the Band structure is not very k-grid sensitive, the DOS depends strongly on the k-grid. A common problem is that of missing DOS: where there are bands there is no DOS, being an artifact of insufficient k-sampling. Using only a better k-grid for the DOS calculation may produce a DOS that is almost as good as if a full calculation (SCF and DOS) was done with the better k-grid. Notice that the effective potential in such a restart with a better k-grid corresponds to the k-grid as was used during the SCF. Therefore the band structure is not affected by using a better k-grid for the restart. See also the RestartDosAndBandStructure example.
This figure shows that the DOS obtained from restarting with a better k-grid is very close to the one obtained with a full calculation with the better k-grid.
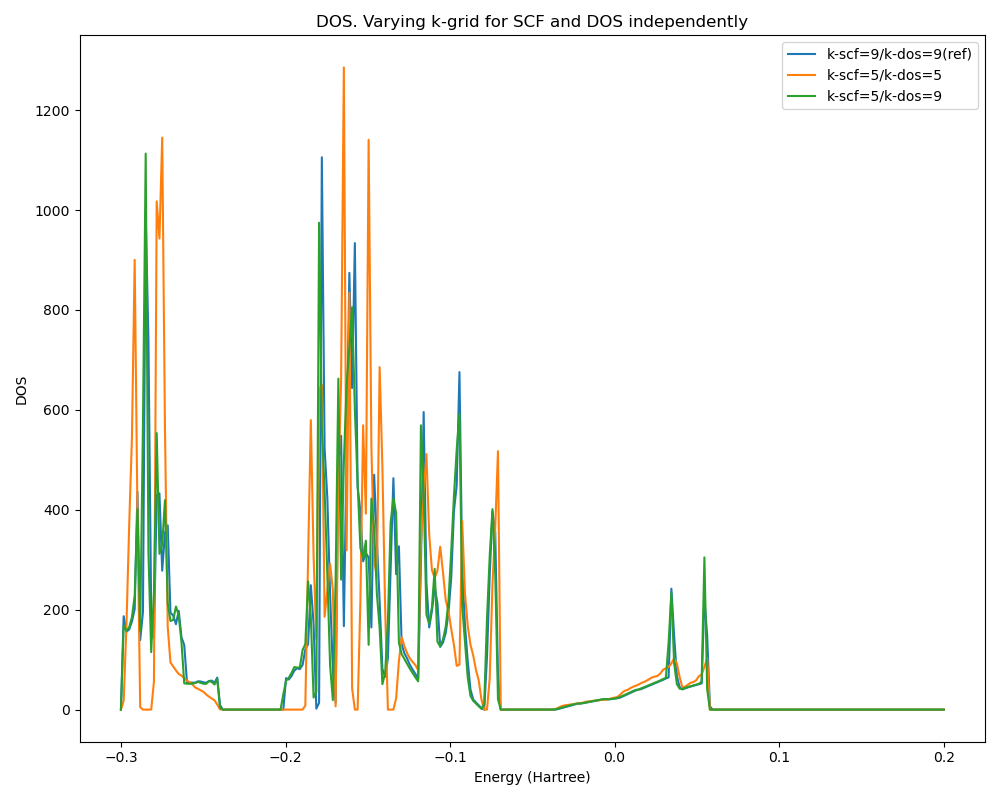
Fig. 8 DOS of a Mo3WSeS7 slab. The best result is when using a 9x9 k-grid for both the SCF and the DOS calculation (blue curve). Using a worse 5x5 grid for both the SCF and the DOS produces a quite different DOS (amber). Doing the SCF with the coarser 5x5 grid and restarting the DOS with the finer 9x9 grid gives the green DOS, matching closely, and mostly hides, the best DOS (blue).¶
Below we show how a missing DOS issue can be solved by either using a better k-grid, or more efficiently by only using a better k-grid for the DOS (using the DOS%Restart option)
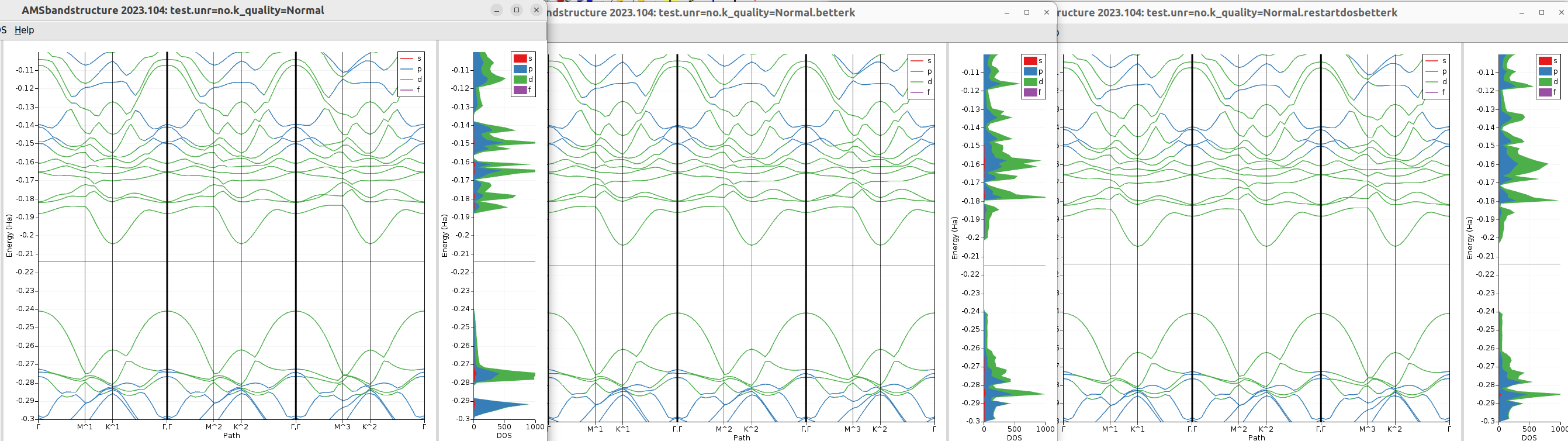
Fig. 9 Illustration of the missing DOS problem for a Mo3WSeS7 slab. Left panel: calculation with a normal k-grid. We clearly see DOS missing in the energy intervals -0.20 to -0.19, and -0.29 to -0.28. Middle panel: using a good k-grid, now the missing DOS appears. The band structure is not much affected, only the lowest band at Gamma being slightly higher with a good k-grid. Right panel: restart from the normal k-grid calculation, but using a good k-grid for the restart. The restarted DOS is very close to the one obtained with the good k-grid for both the SCF and the DOS.¶
While restarts for plotting should be done with the Grid key, the restarting of the DOS/BandStructure should not.