pyCRS : Conformer usage for Database and CRSManager¶
Generating multiple conformers for a compound¶
Different conformers of a molecule can have significantly different sigma profiles, which can lead to big differences in predicted properties with COSMO-RS. For this reason, it’s important for COSMO-RS calculations to use geometries corresponding to the lowest-energy conformer or a set of low-energy conformers when it’s possible several conformers may exist in significant amounts. The script shows how to use the adfcosmorsconformers recipe based on the ConformerTools functionality in AMS to generate a set of low-energy conformers, refine the geometries with a semi-empirical method and finally perform the ADF and COSMO calculations necessary to produce .coskf files.
[show/hide code]
from scm.plams import Settings, init, finish, from_smiles
from scm.plams.recipes.adfcosmorsconformers import ADFCOSMORSConfJob, ADFCOSMORSConfFilter
from scm.conformers import ConformersJob
init()
mol = from_smiles("CC(=O)O")
conf_sett = Settings()
conf_sett.input.AMS.Generator.RDKit
conf_sett.input.AMS.Generator.RDKit.InitialNConformers = 50
conf_job = ConformersJob(name="conformers_uff", molecule=mol, settings=conf_sett)
dftb_sett = Settings()
dftb_sett.input.AMS.Task = "Optimize"
dftb_sett.input.DFTB
# ADFCOSMORSConfFilter(max number of conformers, max energy range)
fil1 = ADFCOSMORSConfFilter(20, 22) # applied to UFF
fil2 = ADFCOSMORSConfFilter(10, 12) # applied to DFTB
fil3 = ADFCOSMORSConfFilter(5, 7) # applied to ADF gas phase
mol_info = {"CAS":"64-19-7", "IUPAC":"Acetic acid", "SMILES":"CC(=O)O"}
job = ADFCOSMORSConfJob(
mol,
conf_gen=conf_job,
first_filter=fil1,
additional=[(dftb_sett, fil2)],
final_filter=fil3,
coskf_name="acetic_acid",
coskf_dir="coskf_acetic_acid",
mol_info=mol_info,
)
job.run()
finish()
Adding conformers to the database¶
This script shows how to add multiple conformers into the database. Please generate the conformers for acetic acid using above script or download the files from the below link before running the below script.
[show/hide code]
from pyCRS.Database import COSKFDatabase
from pyCRS.CRSManager import CRSSystem
import os, glob
################## Note: Ensure to download the coskf_acetic_acid or generating conformers for acetic acid before running the script ##################
conformer_path = os.path.join(os.getcwd(), "coskf_acetic_acid")
db = COSKFDatabase("my_coskf_db.db")
db.add_compound("acetic_acid_0.coskf",coskf_path=conformer_path)
db.add_compound("acetic_acid_1.coskf",coskf_path=conformer_path)
print("Displaying all conformers for acetic acid")
for row in db.get_conformers("Acetic acid"):
print(row)
print("Displaying the conformer with lowest energy structure for acetic acid")
for row in db.get_compounds("Acetic acid"):
print(row)
The output produced is the following
Displaying all conformers for acetic acid
ConformerRow(conformer_id=1, compound_id=1, name='acetic acid', cas='64-19-7', identifier=None, smiles='CC(=O)O', resolved_smiles='CC(=O)O', coskf='acetic_acid_0.coskf', Egas=-1061.593, Ecosmo=-1068.344, nring=0)
ConformerRow(conformer_id=2, compound_id=1, name='acetic acid', cas='64-19-7', identifier=None, smiles='CC(=O)O', resolved_smiles='CC(=O)O', coskf='acetic_acid_1.coskf', Egas=-1056.631, Ecosmo=-1066.71, nring=0)
Displaying the conformer with lowest energy structure for acetic acid
CompoundRow(compound_id=1, conformer_id=1, name='acetic acid', cas='64-19-7', identifier=None, smiles='CC(=O)O', resolved_smiles='CC(=O)O', coskf='acetic_acid_0.coskf', Egas=-1061.593, Ecosmo=-1068.344, nring=0)
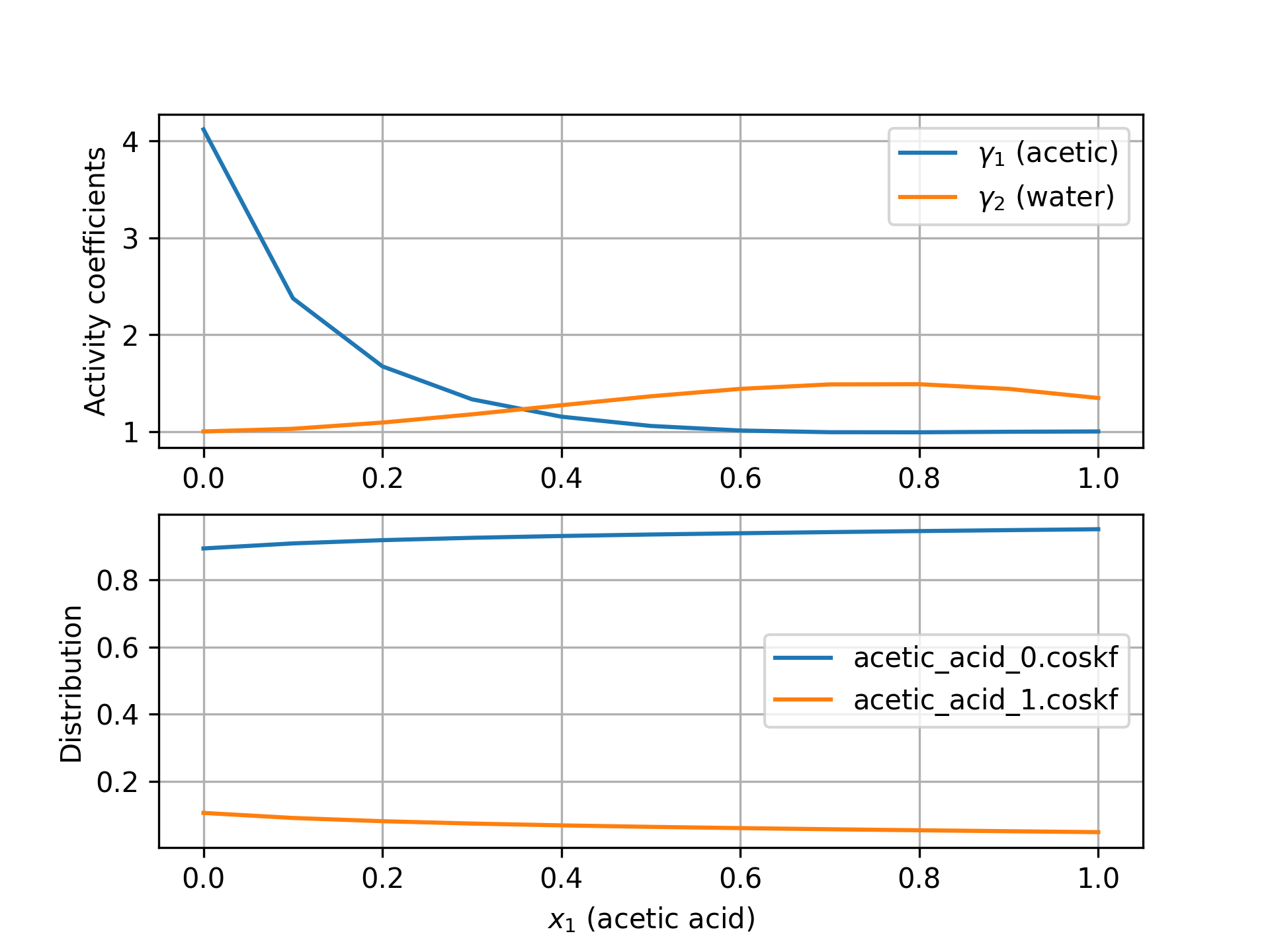
Activity coefficient calculation considering conformers¶
This calculation models acetic acid as a mixture of conformers and plots activity coefficients and the conformer distribution over the mole fraction range. Please generate the conformers for acetic acid using above script or download the files from the below link before running the below script.
[show/hide code]
from scm.plams import Settings, init, finish, CRSJob
from pyCRS.Database import COSKFDatabase
from pyCRS.CRSManager import CRSSystem
import matplotlib.pyplot as plt
import numpy as np
import os, glob
######## Note: Please ensure to install the ADFCRS-2018 database via amspackages (SCM->Packages) before running the below script ########
db = COSKFDatabase("my_coskf_db.db")
db.add_compound("Water.coskf")
System = CRSSystem()
mixture = {}
x_range = np.linspace(0, 1, 11)
for x in x_range:
mixture["Acetic acid"] = x
mixture["Water"] = 1 - x
System.add_Mixture(
mixture, database="my_coskf_db.db", temperature=298.15, problem_type="activitycoef", conformer=True
)
init()
System.runCRSJob()
finish()
gamma_water = []
gamma_acid = []
comp_acetic = {}
for out in System.outputs:
res = out.get_results()
gamma_acid.append(res["gamma"][0])
gamma_water.append(res["gamma"][1])
if comp_acetic == {}:
comp_acetic = out.get_multispecies_dist()[0]
else:
tmp_comp_acetic = out.get_multispecies_dist()[0]
for key in comp_acetic:
comp_acetic[key].extend(tmp_comp_acetic[key])
fig, axs = plt.subplots(2)
axs[0].plot(x_range, gamma_acid, label="$\gamma_1$ (acetic)")
axs[0].plot(x_range, gamma_water, label="$\gamma_2$ (water)")
for struct, val in comp_acetic.items():
axs[1].plot(x_range, val, label=os.path.basename(struct))
plt.setp(axs[0], ylabel="Activity coefficients")
plt.setp(axs[1], ylabel="Distribution")
for i in range(2):
axs[i].legend()
axs[i].grid()
plt.xlabel("$x_1$ (acetic acid)")
# plt.savefig('./pyCRS_activitycoef_conformer',dpi=300)
plt.show()
This code produces the following figure which plots activity coefficients and the distribution of two conformers.