Spin Coupling in Fe4S4 Cluster¶
This ADF tutorial will help you to:
control the spin coupling in multi-center radical systems in two different ways (SpinFlip and ModifyStartPotential),
tweak the SCF convergence in the iron-sulfur cubane case,
visualize the distribution of spin densities in 3D.
Create the Fe4 S4 cubane model¶
Embedded into proteins and coordinated by cysteine ligands, iron-sulfur cubanes are often used by nature in electron transfer and catalysis. While their native structures can be extracted from protein data bank (PDB) files, we will use a model of the Fe4 S4 cubane in this example.
In this step are the instructions to build the system. If you just want the final system go to the end of this step, there you can copy-paste the coordinates.
 → Polyhedra → Cube structure
→ Polyhedra → Cube structureThe cube built is constructed of carbon atoms. We will now change four of these atoms to iron (Fe), and next the remaining four atoms into sulfur (S).
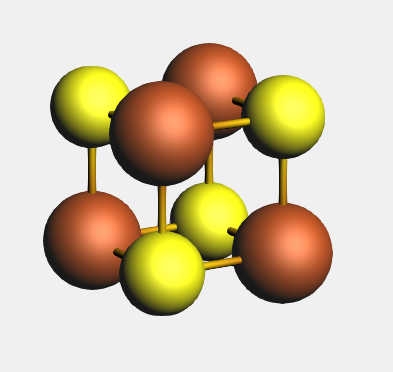
Now you should see the Fe4 S4 cubane in the structure drawing area of AMSinput. The proper coordination to Fe atoms is important in modeling their electronic structure. In proteins, iron-sulfur cubanes are coordinated by cysteine ligands to the Fe sites. Here, we will model these four cysteines by - SH ligands. The procedure to add these ligands is described below.
Tip
The Add Hydrogen command operates on the selected atoms only, if any.
This will replace hydrogen atoms into OH ligands.
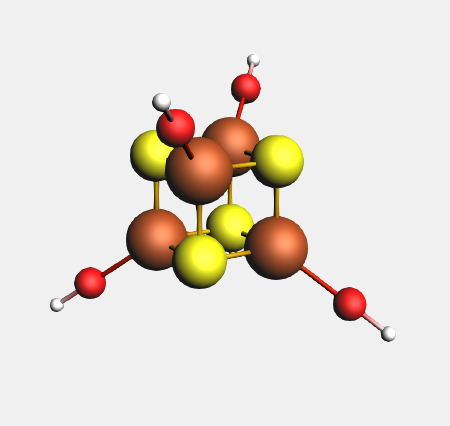

Your Fe4 S4 cubane model should look like this, and have T(D) symmetry:
Instead of building the structure using the instructions above, you can also copy-paste the following coordinates:
Fe 0.92466000 0.92466000 -0.92466000
S 0.92466000 0.92466000 0.92466000
Fe 0.92466000 -0.92466000 0.92466000
S 0.92466000 -0.92466000 -0.92466000
S -0.92466000 0.92466000 -0.92466000
Fe -0.92466000 0.92466000 0.92466000
S -0.92466000 -0.92466000 0.92466000
Fe -0.92466000 -0.92466000 -0.92466000
H 2.61053000 2.61053000 -2.61053000
H 2.61053000 -2.61053000 2.61053000
H -2.61053000 2.61053000 2.61053000
H -2.61053000 -2.61053000 -2.61053000
S -2.03318000 -2.03318000 -2.03318000
S -2.03318000 2.03318000 2.03318000
S 2.03318000 -2.03318000 2.03318000
S 2.03318000 2.03318000 -2.03318000
Optimize the structure with ADF¶
The next step is to optimize this structure. It is a difficult system, and the pre-optimizers will fail. So we will use ADF to optimize the geometry.
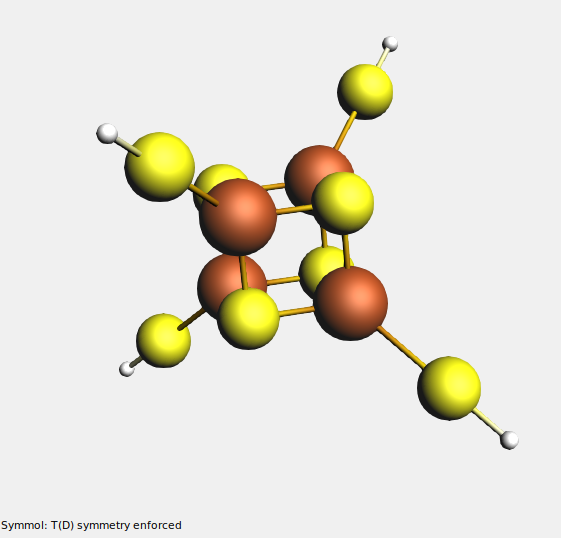
(it should have T(D) symmetry)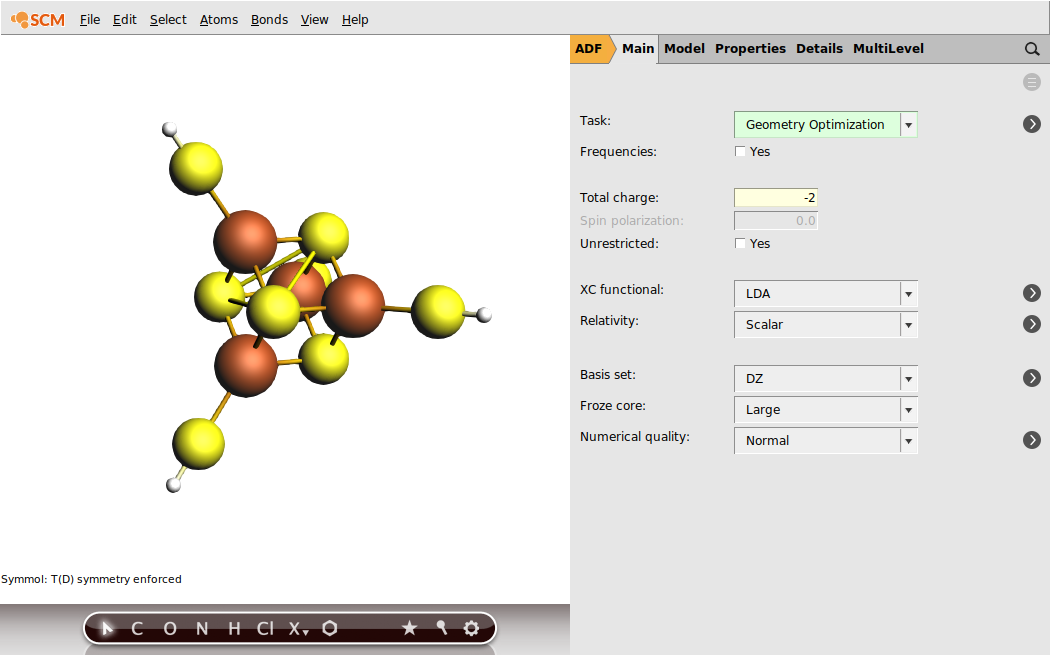
Use AMSmovie to analyze the optimization process in detail:
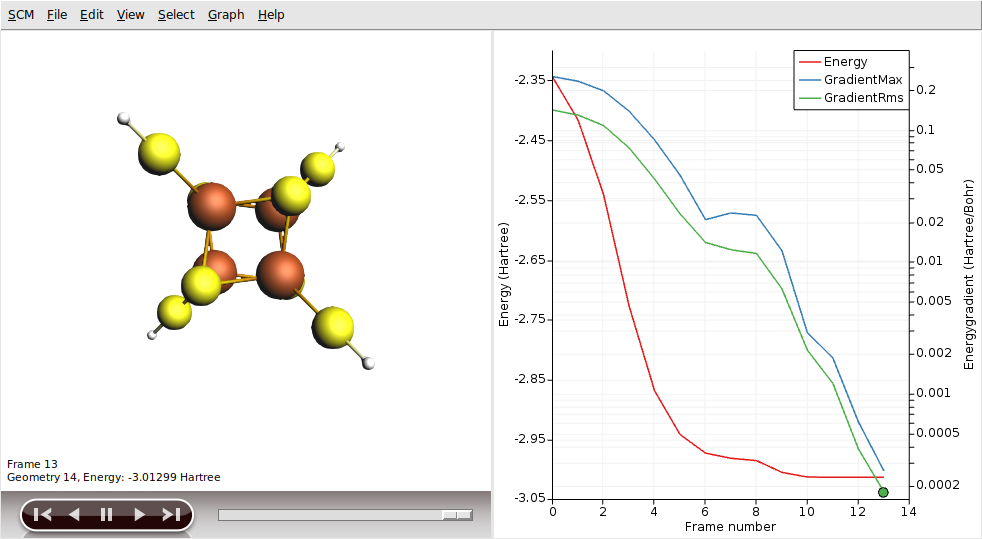
The left panel visualizes the geometry at each step, while the right panel plots metrics like Energy, GradientMax, and GradientRms.
Obtain the solution for the high-spin (HS) state of the cubane¶
In Fe4 S4 systems, the iron sites are commonly high-spin ferrous (Fe3+ , S = 5/2) or ferric (Fe2+ , S = 2). For the present example, we will use the iron-sulfur cubane oxidation state where the two sites are ferric and the remaining two are ferrous. This oxidation level of Fe4 S4 is well-defined and occurs, for example, in rubredoxin and high-potential iron-sulfur proteins (HIPIPs). For our model system, Fe4 S4 (SH)4 , this implies the total charge of -2.
The relative directions, or coupling, of the individual site spin vectors is a very important issue in obtaining the desired density functional solution in Fe4 S4 , as well as many other systems which display a multi-center radical character.
Within the common open-shell approach, the spin vectors are either parallel or anti-parallel. The case when all the spins are parallel is called high-spin (HS). Obtaining self-consisted field (SCF) solution for the HS case is normally simpler because the program does not need to resolve the ambiguity of distribution the sites spin vectors. While the ferromagnetic HS solution commonly does not correspond to the lowest energy electronic state, this solution can be used to obtain the electron density corresponding to the lower energy spin-coupled state. In this step, we will obtain the HS solution for the iron-sulfur cubane, which will be used later in the tutorial. The HS solution for the [Fe4 S4 (SH)4 ]2- model corresponds to S = 2 × 5/2 + 2 × 2 = 9.
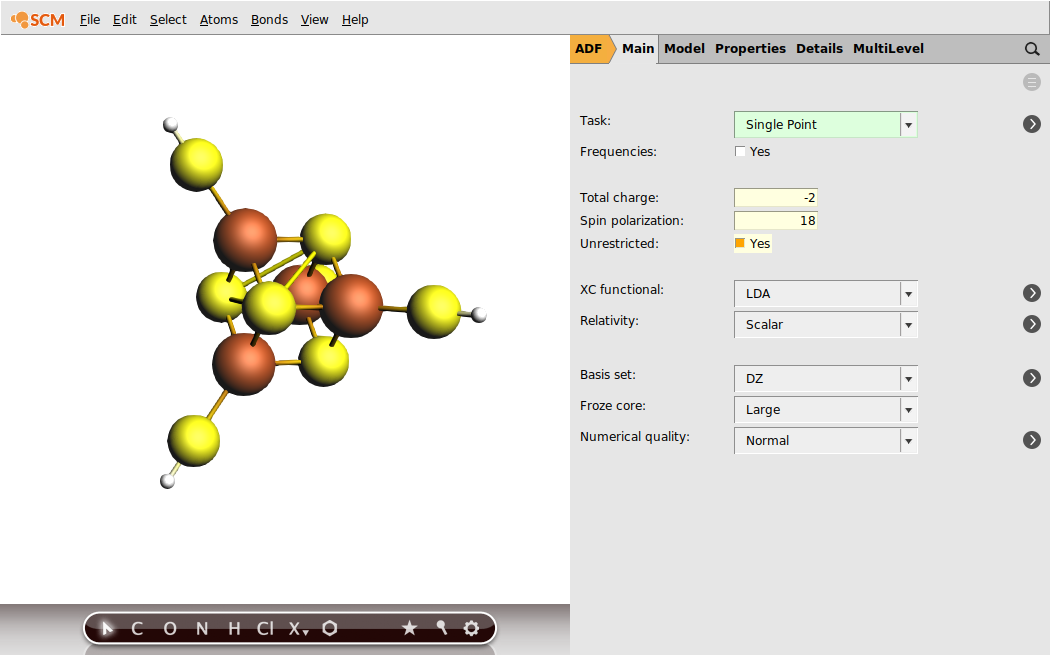
Click on the progress lines so that AMStail window will pop up showing the progress of the job:
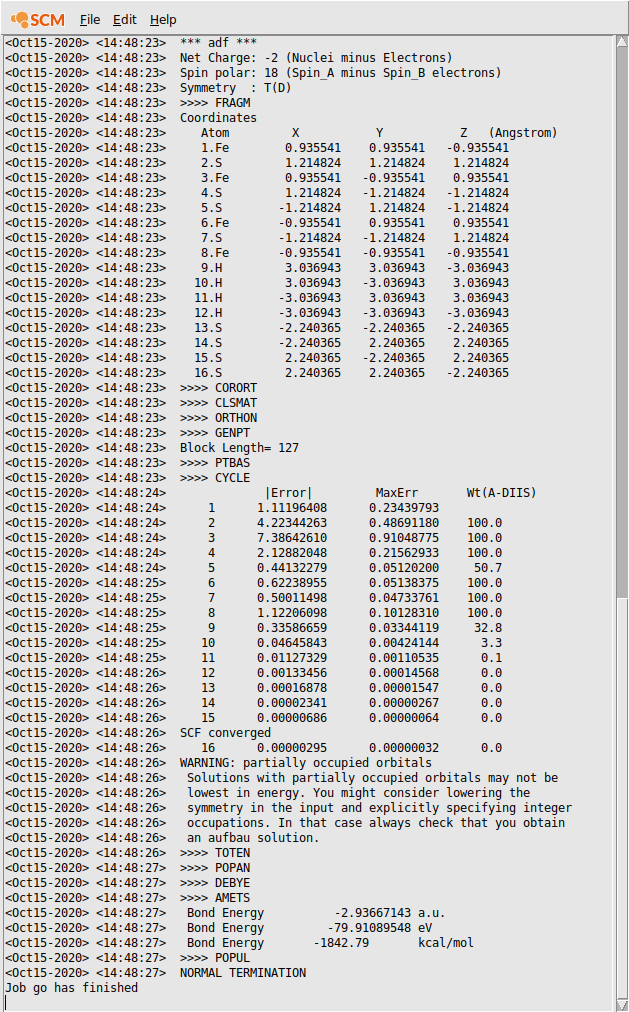
Check in the logfile that the SCF procedure converged.
Couple the spins in Fe4 S4 using the SpinFlip option¶
While SCF solution for the high-spin (HS) state of a multi-center spin system can often be easily found, this solution does not necessarily correspond to the lowest energy state.
To generate the solution with the desired collinear spin arrangement, one option is to use the SpinFlip concept that has been earlier introduced by L. Noodleman and coworkers. In this two step procedure:
first the spin-unrestricted HS solution is generated with all the site spins ferromagnetically coupled in one direction (all spins up, \(\uparrow\) ).
Next the α (\(\uparrow\) ) and β (\(\downarrow\) ) electron densities centered at the sites which are expected to be antiferromagnetically coupled (spins down, \(\downarrow\) ) to the total spin vector are exchanged for the earlier obtained HS solution, using the SpinFlip option, and the calculation is restarted.
Because this approach often results in lowering the electronic symmetry of the system while retaining its structural symmetry, a solution obtained in such way is often called the broken symmetry (BS) state. In such cases you will need to make sure that your BS calculation is done with lower symmetry.
The concept of SpinFlip and BS state can be illustrated considering our iron-sulfur cubane case with two ferrous (Fe3+ , S = 5/2) and two ferric (Fe2+ , S = 2) sites. One of the well-characterized BS states for this level of Fe4 S4 oxidation corresponds to S = (5/2 + 2) - (5/2 + 2) = 0, \(2 \uparrow :2 \downarrow\).
This spin polarization setting corresponds to S = 0 zero spin of the BS electronic state.
The Spin Flip option only works when restarting, so set up the restart calculation from the HS results:
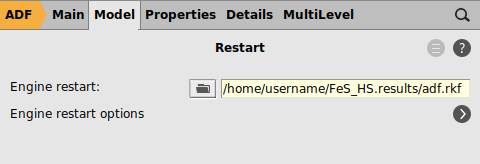
The above will instruct ADF to read the converged HS solution we obtained in the previous step of the tutorial. This solution has been saved in the file Fe_HS.results/adf.rkf.
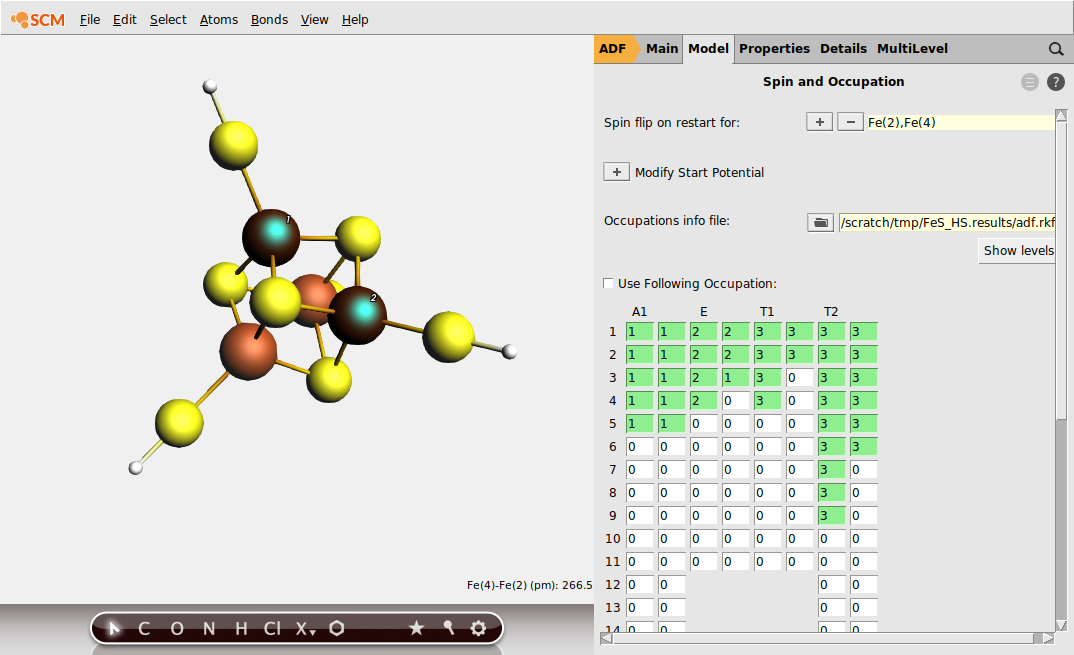
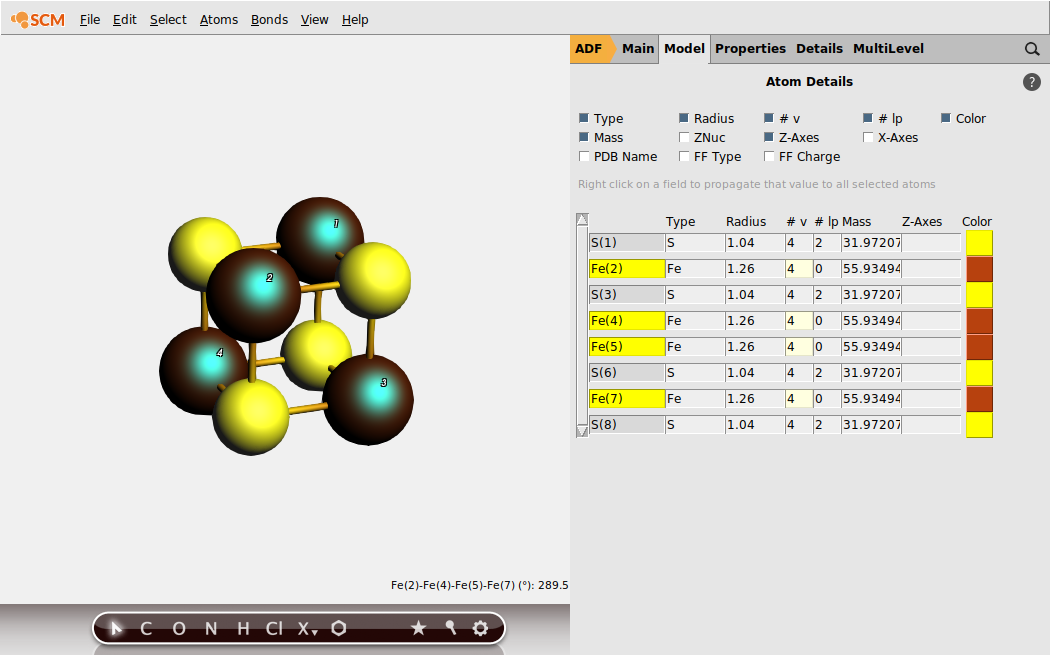
To achieve the desired BS solution, the SpinFlip procedure should be applied to 2 out of 4 Fe sites of Fe4 S4 . In the above example, we selected sites Fe(2) and Fe(4). This will instruct SpinFlip algorithm to interchange α (\(\uparrow\) ) and β (\(\downarrow\) ) electron densities associated with these two Fe sites when the job will be restarted, changing the HS \(4 \uparrow :0 \downarrow\) spin state to the BS \(2 \uparrow :2 \downarrow\) spin state.
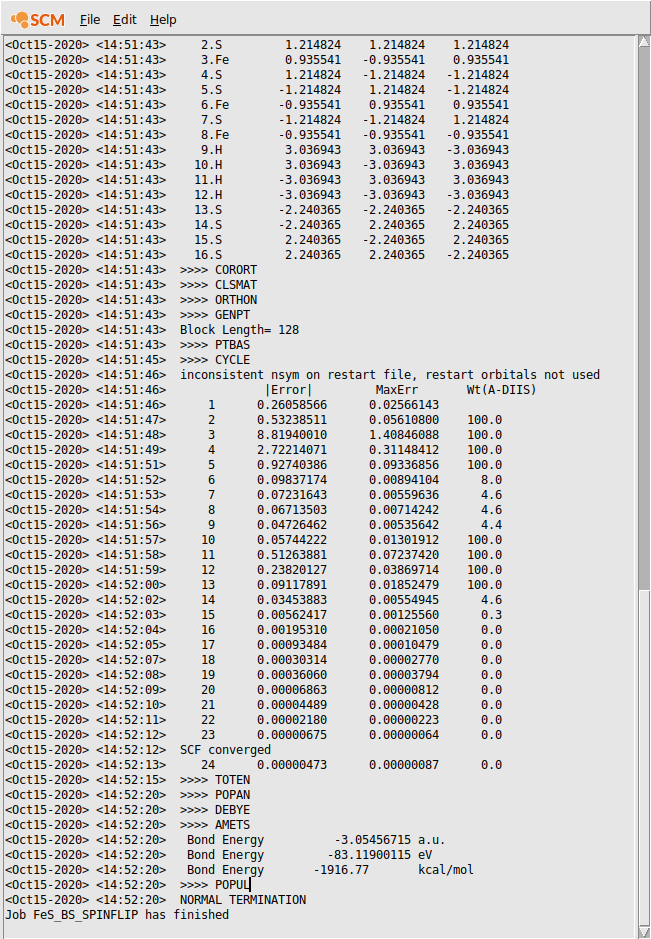
Check in the logfile that the SCF procedure converged.
Coupling the spins using the ModifyStartPotential option¶
There is an alternative to SpinFlip available in ADF, which is aimed to achieve a specific spin-coupled solution in a single calculation only. This is done using the MODIFYSTARTPOTENTIAL key in ADF: it allows you to create a spin-polarized potential at the very start of the calculation.
Please read the page of ADF manual on the MODIFYSTARTPOTENTIAL key prior to proceeding with this step of the tutorial. As follows from the MODIFYSTARTPOTENTIAL description, this key allows to control the ratio of spin-α and spin-β electrons associated with fragments via ‘alpha’ and ‘beta’ numbers. For the purpose of the present tutorial, we will consider the four Fe sites as fragments. ‘Alpha’ and ‘beta’ numbers will correspond to the number of spin-α and spin-β electrons, respectively, associated with a Fe site.
So follow these instructions to obtain the BS solution via the MODIFYSTARTPOPTENTIAL option:
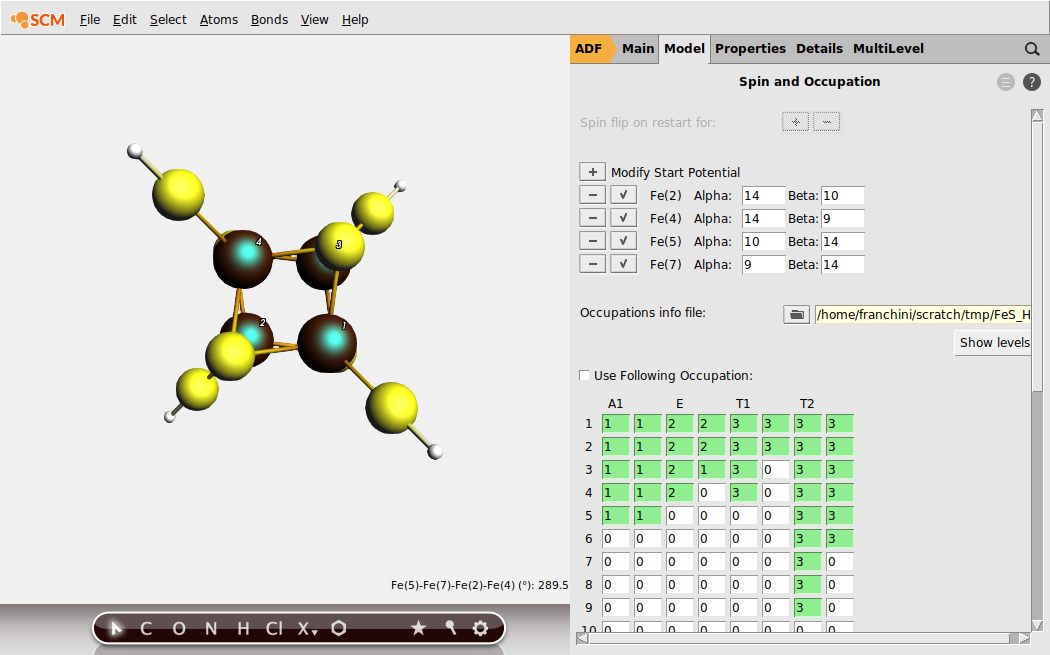
For the spin-up Fe3+ , S = 5/2, the alpha and beta numbers would be 14 and 9, correspondingly, and for the spin-up Fe2+ , S = 2, these numbers are 14 and 10. For the spin-down Fe sites, alpha and beta numbers should be apparently transposed. Also, our desired BS state for this level of Fe4 S4 oxidation corresponds to S = (5/2 + 2) - (5/2 + 2) = 0, \(2 \uparrow :2 \downarrow\).
Note that SCF procedure might be problematic.
You can play with several options to help the convergence. In the SCF panel (in Details) you can experiment with several methods like LISTi, A-DIIS, E-DIIS, ARH and the option to enable the old SCF algorithm. In the SCF convergence details panel further options are available, like mixing, level shifting, orbital freezing and DIIS details.
In this particular case, the default method works but it needs a lot of iterations. So no need to make changes.
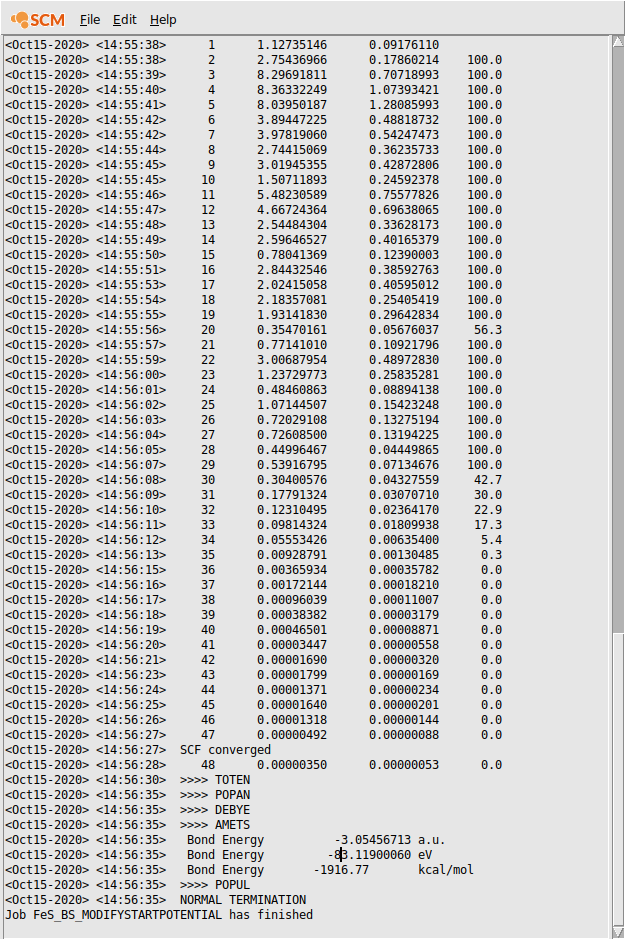
Hopefully you will be able to converge the job, to the same energy and state as the SpinFlip job.
Thus both the SpinFlip and the ModifyStartPotential option allow you to obtain a desired Fe spin coupling pattern in the Fe4 S4 case. While SpinFlip is a two-step approach and ModifyStartPotential works as in a single step, the SpinFlip approach shows a better performance during SCF (much better and faster SCF convergence).
View the spin density of the broken symmetry (BS) solutions¶
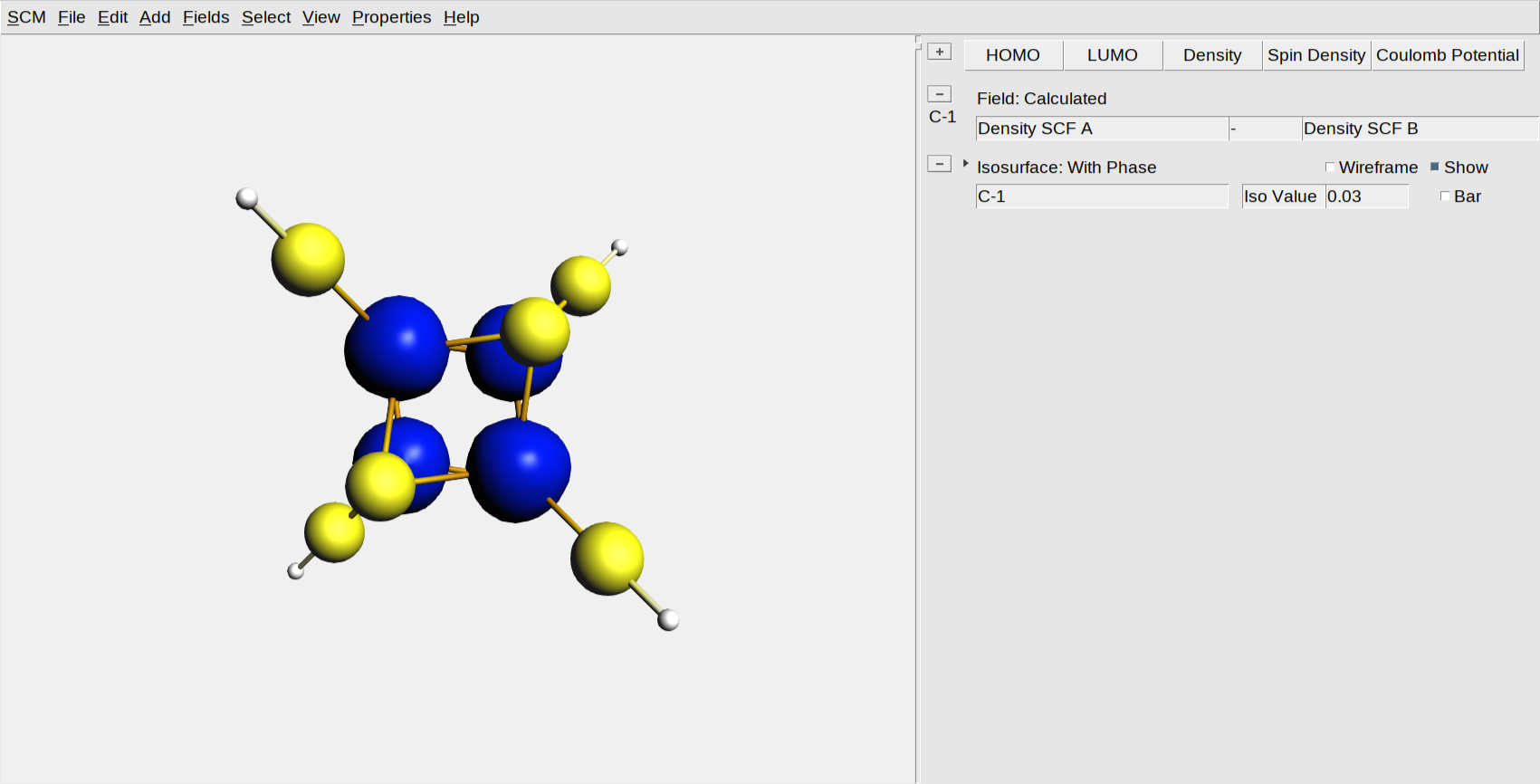
In the two previous steps of this tutorial, we have generated the broken symmetry (BS) solution for the Fe4 S4 cubane using alternatively the SpinFlip and ModifyStartPotential options. Here, we will analyze this BS solution viewing the Fe spin densities using AMSview, and confirm that the spin alignment of the iron sites is \(2 \uparrow :2 \downarrow\). This type of analysis can also be useful for presentations and scientific illustrations.
You should see the [Fe4 S4 (SH)4 ]2- system in the AMSview window.
The spin-density is available as a short-cut in the Properties menu:
You should see now the two Fe ions surrounded by blue blobs (spin up, \(\uparrow\) ) and the other two by red blobs (spin down, \(\downarrow\)):
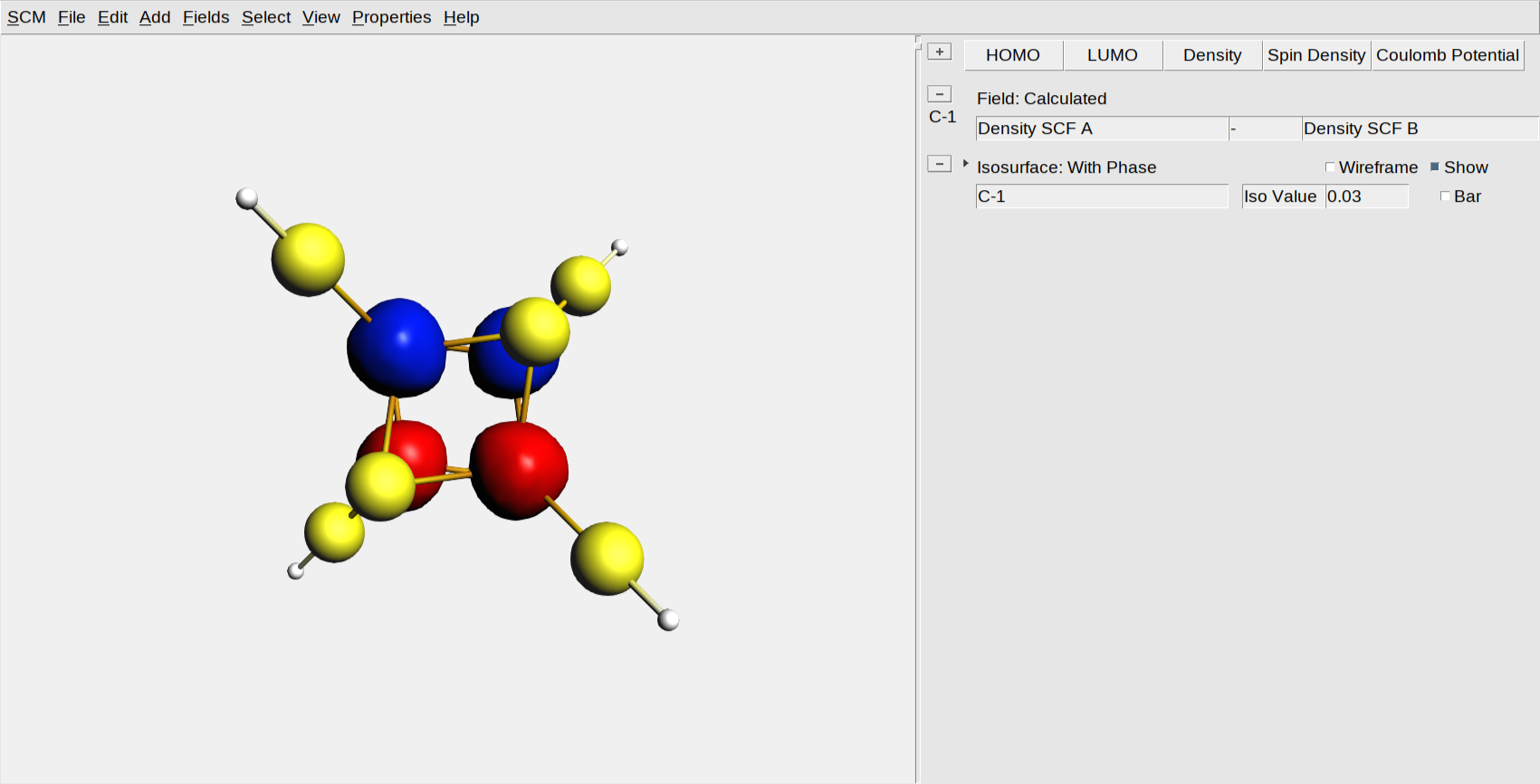
Note that you now have two visualization lines: one is a calculated field that actually calculates the difference between the alpha and beta spin density. The other is an isosurface with phase that visualized the calculated field. You can use these calculated fields for many purposed. For example, by changing the ‘-’ into a ‘+’ you are visualizing the total density instead of the spin difference density.
In the same way you can also check the spin densities for the FeS_HS and FeS_BS_SPINFLIP results. The FeS_BS_SPINFLIP should look the same as the FeS_BS_MODIFYSTARTPOPTENTIAL, the FeS_HS should look like: