Work functions at interfaces¶
This tutorial will show how to use BAND to calculate the work function Φ of the
Al(100)/vacuum interface
Al(100)/LiF(100) interface
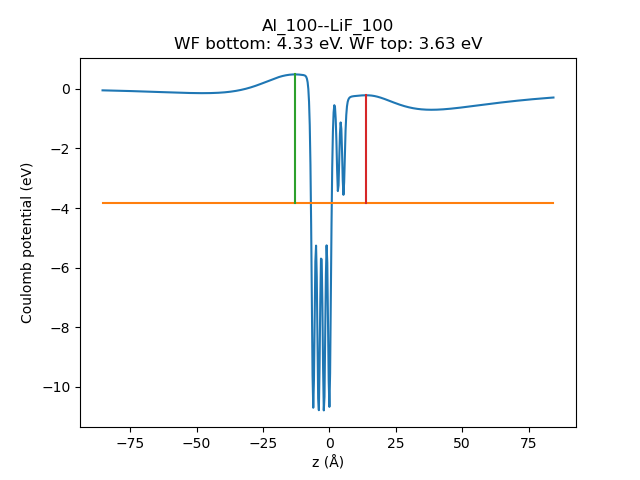
Fig. 47 Plane-averaged electrostatic (Coulomb) potential vs z coordinate. The orange horizontal line is the Fermi energy. The red and green vertical lines indicate how the work function is calculated on either side of the slab. The work function (WF) for the Al(100)/vacuum interface (green) is 4.33 eV. The WF for the Al(100)/LiF(100) interface (red) is 3.63 eV.¶
In the above figure the work function Φ is calculated as
Φ = local maximum in Coulomb potential - Fermi energy
The adsorption of LiF(100) will decrease the work function, as compared to vacuum. The results will be compared to plane-wave-DFT results by Prada S., et al. [1] and Kondo and Matsushista [2]
Al(100) Φ [eV] |
Al(100)/LiF(100) ΔΦ [eV] |
functional |
code |
|
Prada et al. |
4.37 |
-0.7 |
PW91 |
VASP |
Kondo and Matsushista |
4.28 |
-0.59 |
PBE |
Quantum ESPRESSO |
This tutorial |
4.33 |
-0.70 |
PW91 |
BAND |
Note
The above works differ not only in code and functional, but also basis set, k-point sampling, number of layers in the Al or LiF slabs, whether the system is relaxed or not, …
See also
Work function example with Quantum ESPRESSO
Build the initial system¶
 →
→ 
When setting up a solid-solid interface, the lattice constants must match. Both Al and LiF are cubic, so if their lattice constants match, also the (100) surface lattice constants will match. At least one of the materials needs to be a bit strained. Here, we will strain the LiF slab to match the Al slab. We will place the LiF slab at a distance of 3.27 Å from the Al slab.
Note
For other interfaces, you may need to apply surface rotations and use surface supercells if the lattice constants of the two materials are very different, in order to not apply too much strain to one of the materials.
Download the LiF-on-Al.xyz file
Select File → Import Coordinates and select the downloaded file
See also
If you are not yet familiar with the editing tools in AMSinput, take a look at our Introduction to Building Structures.
First, build the Al(100) slab:
2
Similarly, open a new AMSinput window and create the LiF(100) slab:
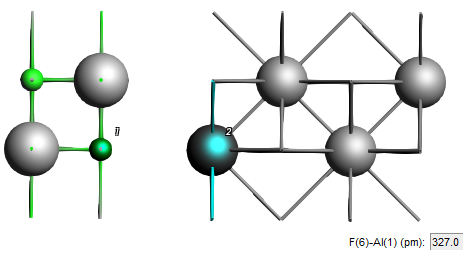
We now want to position this LiF slab on top of the Al slab such that the F atom is 3.27 Å directly above the top Al atom
3.27 Å to first atom (the first atom is the first selected F atom)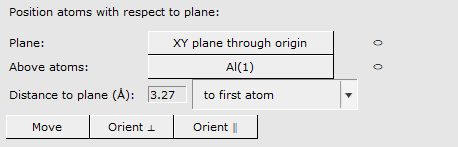
This has created the desired interface geometry.
Set up the DFT calculation¶
Then, set the BAND settings:
 next to K-space
next to K-space7 7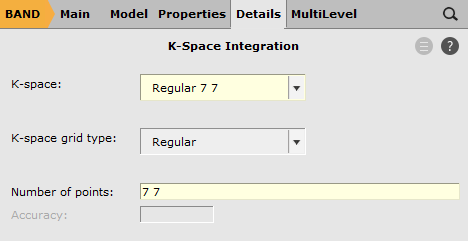
0.002 hartree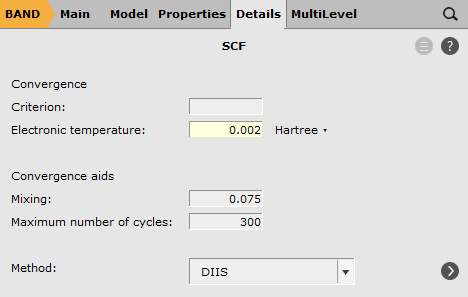
This sets up a 7x7 k-space grid. The integration, spline zlm fit, and electronic temperature options help with the SCF convergence.
Save and run the job.
Get work function with Python script¶
WorkFunctionVacuumAndInterface.pyresults_dir variable to give the correct path to the .results folder from the previous calculation. It should contain two files: ams.rkf and band.rkf.$AMSBIN/amspython WorkFunctionVacuumAndInterface.pyThis produces a plot like the below:
Fig. 48 Plane-averaged electrostatic (Coulomb) potential vs z coordinate. The orange horizontal line is the Fermi energy. The red and green vertical lines indicate how the work function is calculated on either side of the slab. The work function (WF) for the Al(100)/vacuum interface (green) is 4.33 eV. The WF for the Al(100)/LiF(100) interface (red) is 3.63 eV.¶
Get work function with the graphical user interface¶
100 Å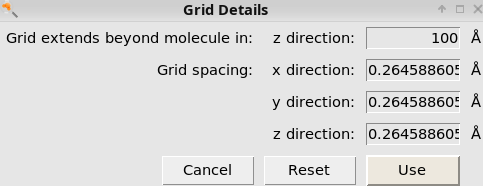
Then, switch to the line graph generator:
01000This produces the following plot:
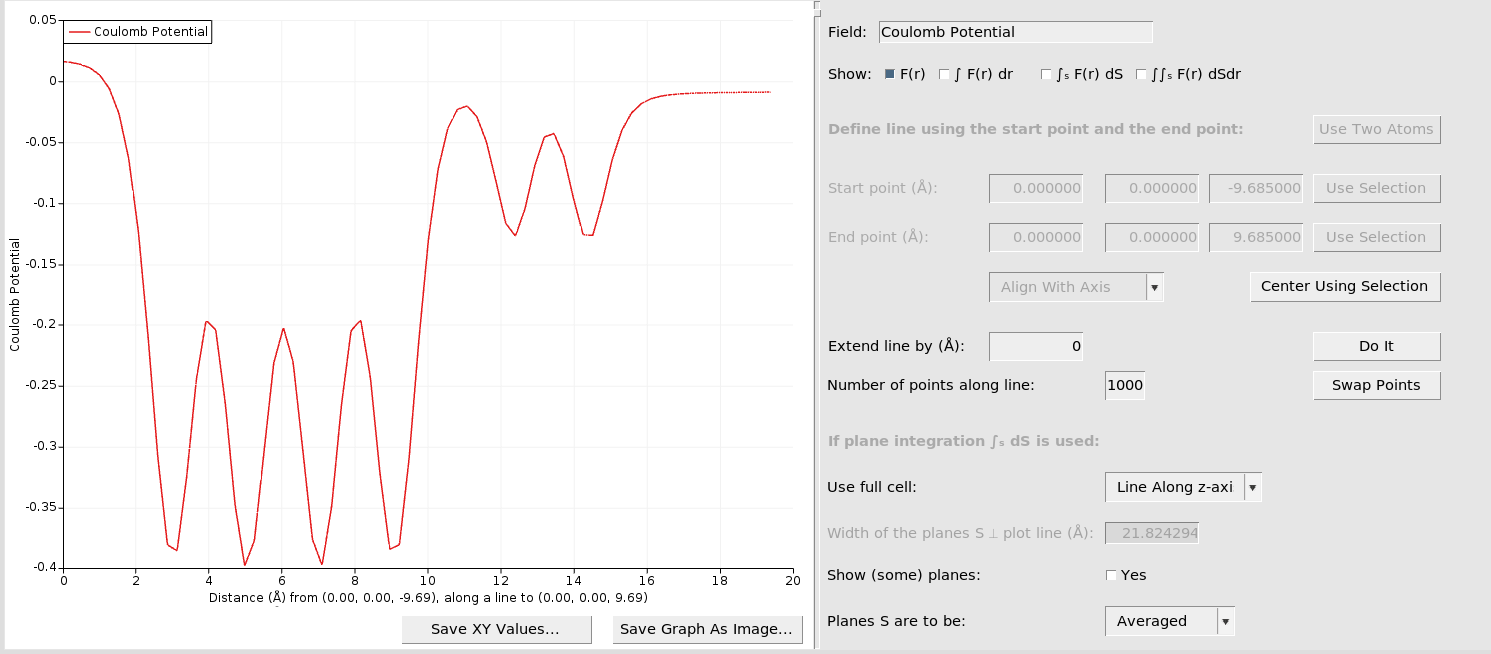
To extend the plotted potential further out into the vacuum:
80Note that the extension which here is set to 80, which needs to be smaller than the “Grid extends beyond molecule” previously set to 100.
This should produce a figure like the following:
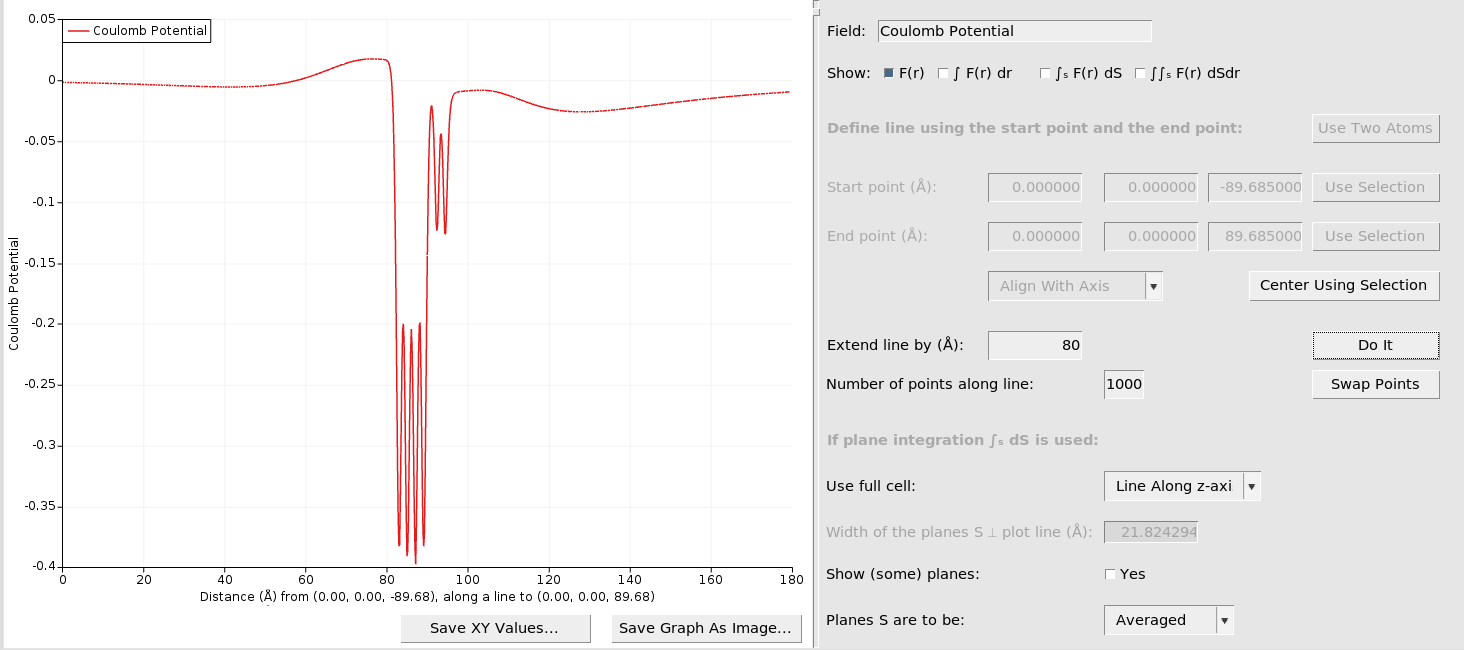
If you hover over the first and second local maxima at the left hand-side, you can read off the y coordinates as
left-hand side (bottom side of slab, Al/vacuum): 0.0176 hartree
right-hand side (top side of slab, Al/LiF): -0.0081 hartree
These plane-averaged potentials should be compared to the Fermi energy to get the work function.
This gives:
Band gap information
No band gap
Fermi Energy: -0.1413 a.u.
Fermi Energy: -3.8443 eV
Consequently, we get
bottom side Φ = 0.02555 - (-0.1413) = 0.16685 hartree = 4.33 eV
top side Φ = -0.0081 - (-0.1413) = 0.1332 hartree = 3.62 eV