Getting started: Geometry optimization of ethanol¶
This tutorial will help you to:
create a simple molecule
view the molecule from all sides and save a picture
make a couple of changes to the molecule with different tools
set up your ADF calculation
perform the actual ADF calculation
visualize some results: energy levels, geometry, electron density, orbitals, …
See also
More detailed information on the features presented in this tutorial can be found in the AMS driver manual and ADF manual.
Step 1: Preparations¶
Start AMSjobs¶
On a Unix-like system, enter the following command:
On Windows, one can start AMSjobs by double-clicking on the AMS-GUI icon on the Desktop:
On MacOS, use the AMS2025.xxx application to start AMSjobs:
Note that the directory in which AMSjobs depends on how you start AMSjobs, so your screen might look different.
Make a directory for the tutorial¶
We prefer to run the tutorial in a new, clean, directory. That way we will not interfere with other projects. AMSjobs not only manages your jobs, but also has some file management options. In this case we use AMSjobs to make the new directory:
Start AMSinput¶
Now we will start AMSinput in this directory using the SCM menu:
The AMSinput module should start:
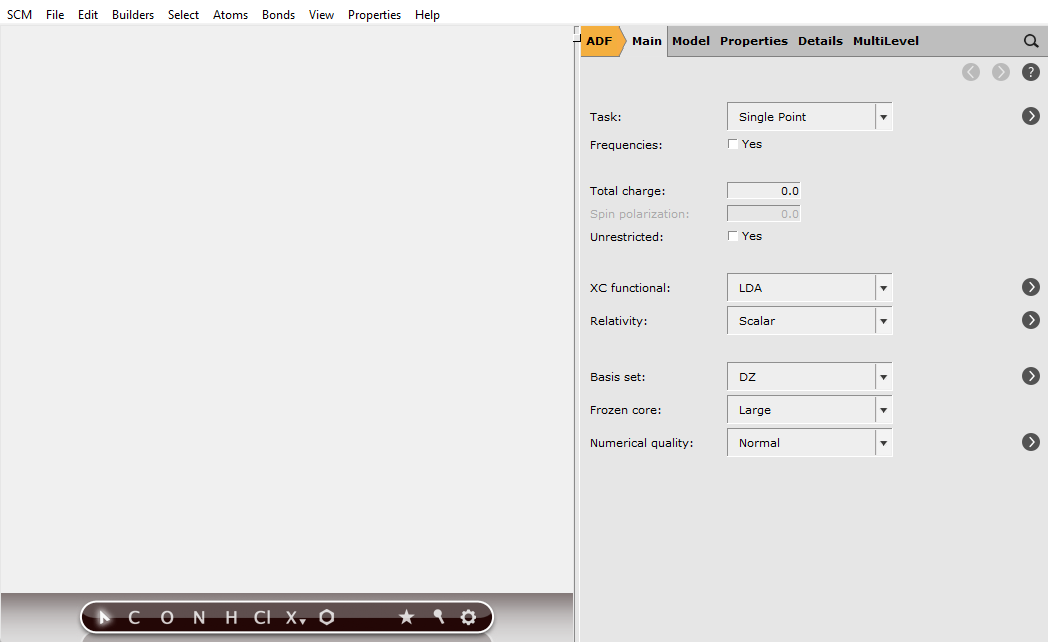
The AMSinput window consists of the following main parts:
the menu bar with the menu commands (SCM, File, Edit, …). On a Mac the menu bar is at the top of the screen
the drawing area of the molecule editor, on the middle left side
the status field at the lower part of the drawing area (blank when AMSinput is empty as shown above)
the molecule editor tools, below the drawing area
many panels for input options, on the right side. Currently the ‘Main’ panel for ‘ADF’ is visible
Panel bar with menu commands to activate the module and options panel of choice
a search tool

Undo¶
AMSinput has an Undo command (Edit → Undo), which works on your molecule (thus not on your input options).
If you make a mistake while making changes to your molecule, just use the Edit → Undo menu command to go back in time. You can Undo more than one step, or Redo a step (with Edit → Redo) if you wish to do so.
Step 2: Create your molecule¶
Create a molecule¶
The molecule we are going to create is ethanol.
First we will draw the two carbon atoms, next the oxygen atom, and after that we will add all hydrogen atoms at once. Finally, we will pre-optimize the geometry within AMSinput.
Create the first carbon atom¶
To create an atom, you need to select an atom tool.
Back glow is added to the ‘C’ button to indicate that you are using the C-tool. Also, the status field in the left bottom corner shows ‘C tool’ to indicate that you are using the C-tool.
Now create the first carbon atom:
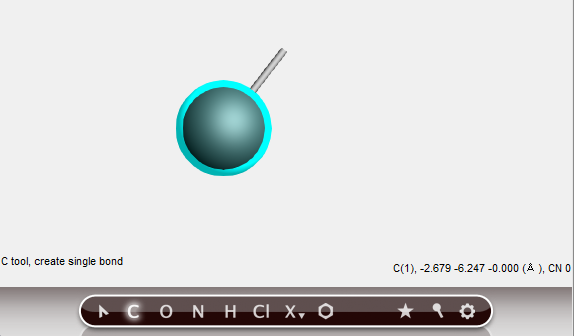
One carbon atom has been created.
Note that:
if you move the mouse you will see a white line from that carbon atom to the current mouse pointer position: this shows you are in ‘bonding’ mode, and that the bond will be made to the atom just created
the ‘C’ button has back glow, indicating you are still using the C-tool
the cyan halo around the carbon atom indicates that it is the current selection
the status field contains information about the current selection: it is a Carbon, number 1, its coordinates and the current coordination number (number of bonds)
the status field also shows the current tool (C), and that a single bond will be made
Create the second carbon atom¶
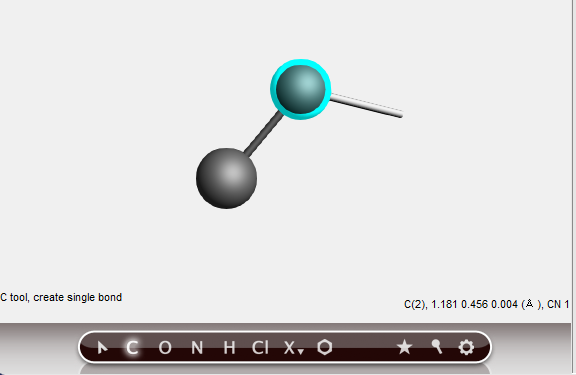
A second carbon atom has been created, bonded to the first atom.
The atom will be created along the ‘bonding line’, at a distance that corresponds to a normal C-C single bond distance. That is, the bond length is constrained while drawing.
The newly created atom becomes the new selection, still using the C-tool in bonding mode. The next bond will be created to the last atom added.
Create the oxygen atom¶
To create the oxygen atom you need to switch to the O-tool:
With the O-tool, create an oxygen atom bonded to the second carbon;
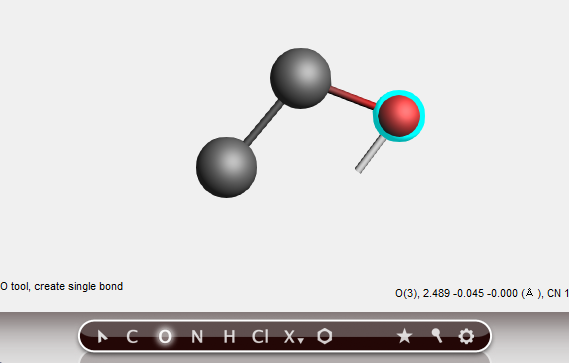
The oxygen atom has been added.
For now, we are done using atom tools, so go back to the select tool:
Add the hydrogens¶
Now many hydrogen atoms need to be added. You can do this using the H-tool, but a much easier method is to use the Atoms → Add Hydrogens menu command:
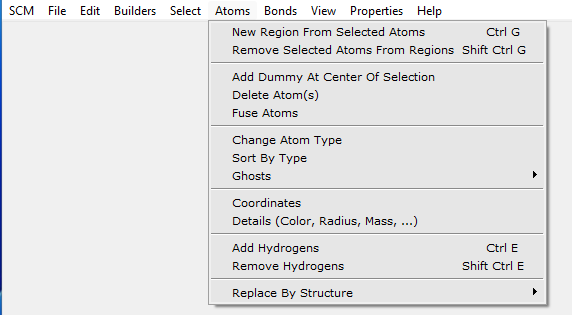
The ‘Add Hydrogen’ menu command works on the selection only, when present. Thus, only one hydrogen atom would be added to the oxygen atom. This is not what you want. So first we make sure that nothing is selected by clicking in empty space.
Now no atoms are selected any more.
Many menu commands have shortcuts. In this case you can also use the shortcut (ctrl-E or cmd-E, depending on your platform) as an alternative. The shortcuts are indicated in the menu commands.
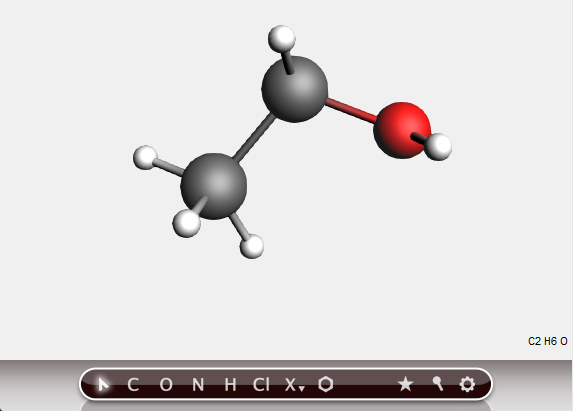
All atoms will be saturated with hydrogen atoms. And you have created an ethanol molecule, though the geometry is still far from perfect.
Pre-optimize the geometry¶
Now use the optimizer that comes with AMSinput to pre-optimize the geometry.

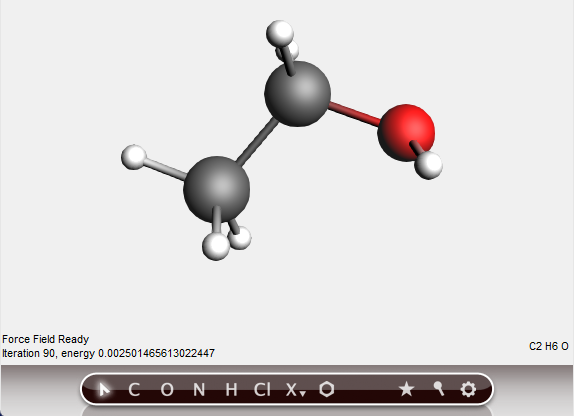
The geometry of the molecule will be pre-optimized, using the Universal Force Field (UFF) by default.
Tip
You can change the default method for pre-optimization via SCM → Preferences, or use a different pre-optimizer by right-clicking on the cog wheel and selecting the method to use from the pop-up menu.
In the status field below the drawing area you can follow the pre-optimization iteration number and the energy relatively to the starting configuration.
Viewing the molecule¶
Rotate, translate, or zoom¶
You can rotate, translate, and zoom your molecule using the mouse.
You need to drag with the mouse: press a mouse button, and while holding it down move it. Which mouse button, and which modifier key you press at the same time, determines what will happen:
Rotate |
Left |
Rotate in-plane |
ctrl-Left |
Translate |
Right |
Zoom |
Mouse wheel, or (not on windows) alt-Left (drag up or down) |
The rotate, translate, and zoom operations change how you look at the molecule, they do not change the coordinates.
This behavior is the default behavior, you can change what the right mouse button does using the Preferences.
Save picture¶
You can save a picture of your molecule using the ‘Save Picture …’ command from the File menu.
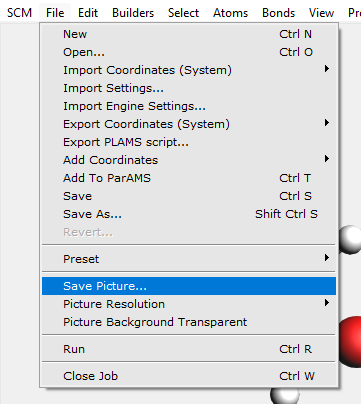
A picture will be saved to disk containing the figure of your molecule. Only the drawing area is saved in the picture, not all the input.
Tip
The default format used is PNG. You can change this in SCM → Preferences. You can also change the resolution via File → Picture Resolution. A smaller resolution will result in a smaller file, but will reduce the quality.
Molecular conformation¶
Rotate such that you look along the C-C axis¶
Select the top CH3 group¶
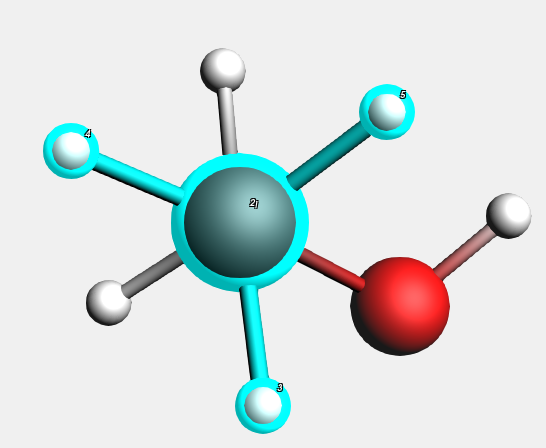
As you will notice, all atoms directly connected to the selected atom are added to the selection. Alternatively, you can also make a selection by shift-clicking on the elements you want to select.
This has almost the same effect (in this case you have not selected the second carbon atom).
Rotate the selection¶
We are now trying to make an eclipsed geometry.
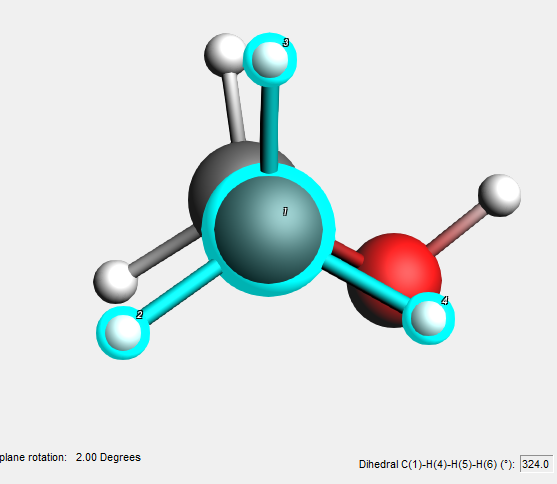
You can move the selection by clicking in a selected object, and dragging with the mouse. All usual operations are possible: rotate, rotate in-plane, translate and zoom. Zoom in this case means moving the selection perpendicular to the screen. If you click in empty space, instead of in a selection, you will manipulate the whole molecule instead of the selection.
In the status field you see the current rotation angle.
Back to Staggered Geometry¶
The optimizer will bring the structure back to the original staggered geometry. If it does not complete, repeat this step until it does.
Getting and setting geometry parameters¶
Bond length¶
First select the oxygen atom and the connected hydrogen atom.
This time we make the selection by dragging a rectangle around all objects that we want to select.
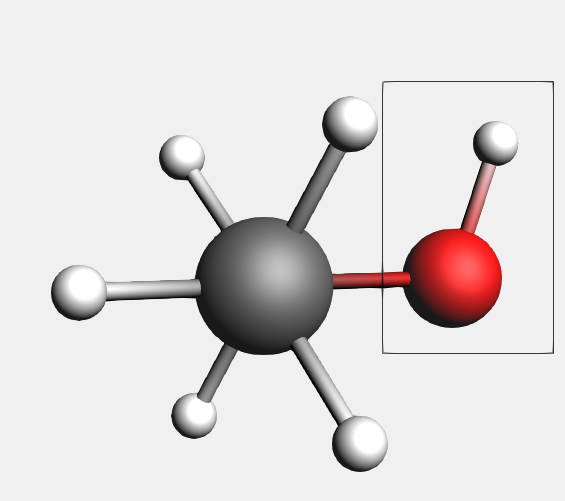
The oxygen atom and the hydrogen atom are selected.
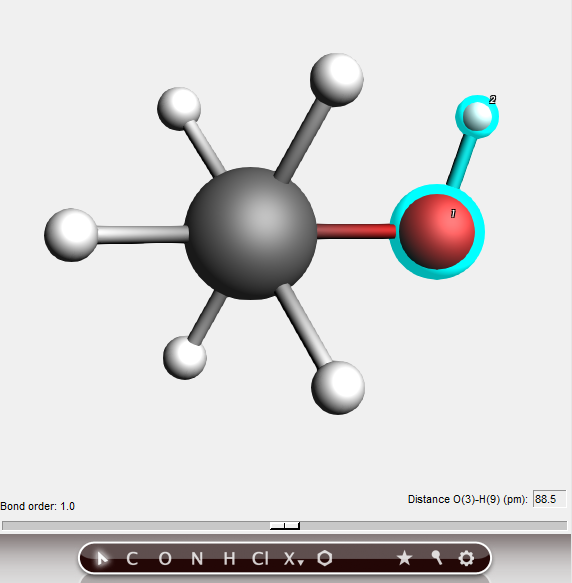
In the status area you see the distance between the selected atoms, information about the bond, and a slider.
You can set the distance to any value you wish by editing it in the rectangle showing the current value, or by using the slider.
Tip
The order in which you have selected the atoms is shown with numbers. By default, the last atom selected will will be in the group of atoms to move.
Press the control key while using the slider, and the smallest group of atoms will move.
Bond angle¶
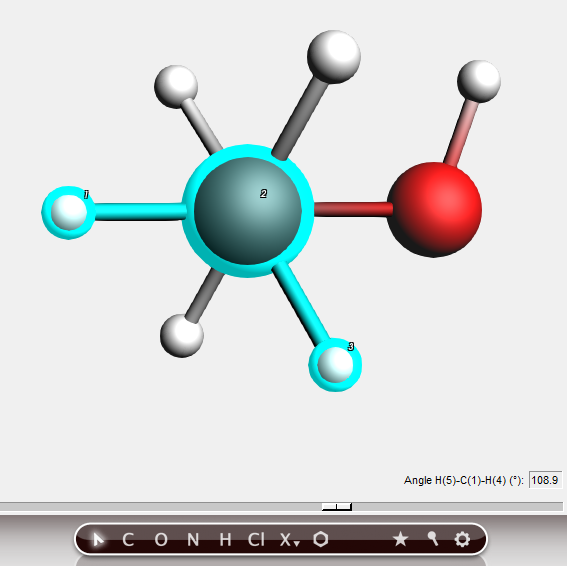
In the status area information about the bond angle of the selected three atoms is given, and the slider is again visible. You can change this value to a value you like by editing the value field or using the slider.
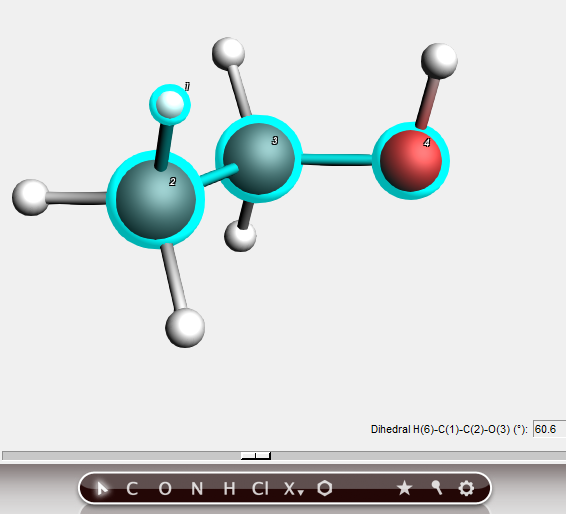
Dihedral angle¶
By selecting four atoms we get information about the dihedral angle. And of course you can also change it, most conveniently using the slider.
Coordinates¶
To view the coordinates we have to go to a different input panel. The input panels can be selected using the panel bar on the top of the input panels, the right half of the window.
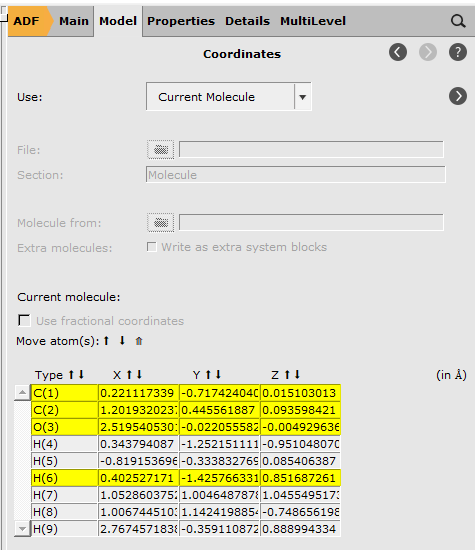
You get a list of all Cartesian coordinates. They will be updated in real time when you make changes to the molecule, and you can also edit the values yourself. In that case, the picture of the molecule will be updated automatically.
Note that some atoms are highlighted. These are the currently selected atoms.
The Move Atom(s) buttons (the arrows) will move the selected atoms up or down. In this way you can re-order the atoms.
Extending and changing your molecule¶
Before making some changes, let’s re-optimize. We first select the ‘Main’ panel so the coordinates will not be visible during the pre-optimization. Otherwise this may slow down the pre-optimization.
Let’s try to change the CH2OH group in a COOH group.
Thus, we need to:
remove one hydrogen
change one hydrogen into an oxygen
change a single bond into a double bond
After this, we will revert to the ethanol molecule.
Delete an atom¶
First: delete one hydrogen
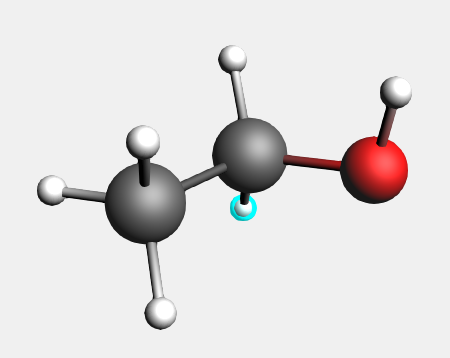
The selected atom is removed.
Change the type of an atom¶
Next, we will change a hydrogen into an oxygen atom.
Tip
You can quickly select a tool using the C, O, H, N, S, P, or F keys.
Or use X, type one or two letters, and Return, for any element.
Change the bond type of an existing bond¶
Now the oxygen atom is singly bonded to the carbon, we need to change this into a double bond.
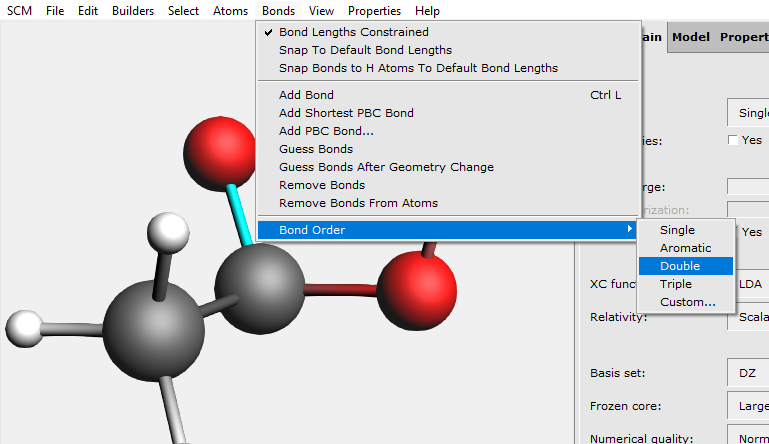
The single bond has changed into a double bond.
Another way to modify a bond type is to click on the bond once which will select this bond. Then click on the bond tool in the molecule editor panel (‘ball and stick’ logo to the right of the start), and select the proper bond type.
Tip
Use the keyboard shortcuts: select a bond, press 1 for a single bond, 2 for a double bond, 3 for a triple bond and 4 for an aromatic bond.
To get a reasonable geometry optimize the structure:
Add new (bonded) atoms¶
Now, to revert to the ethanol molecule, we first remove the new doubly-bonded oxygen atom, and then add one hydrogen atom.
Note that this way you started bonding mode again, as indicated by the bond to the mouse position.
For the next step, we want to find the optimal geometry of a local minimum, for this
Make sure the O-H bond remains parallel to the C-C bond, or repeat this process.
Warning
The results in the next section may look slightly different for other energy minima.
Once again you have created an ethanol molecule.
Tip
When all atoms or none are selected, the pre-optimizer will be applied to the entire system. You can also choose to optimize part of your system by selecting a subset and applying the pre-optimizer only to that selection when prompted.
Step 3: Select calculation options¶
Task¶
AMS has many different modes of operation, or tasks.
See also
Tasks section of the AMS driver manual.
To optimize the geometry of the ethanol molecule we choose the geometry optimization task:
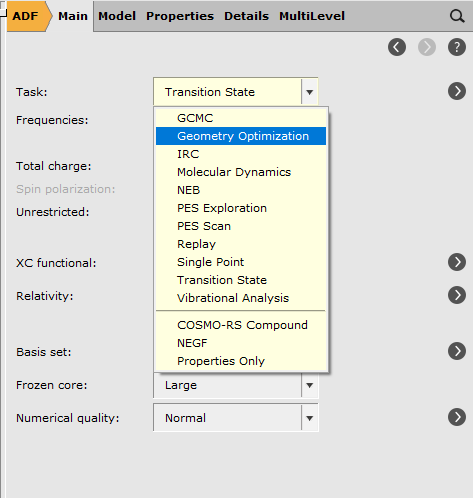
After selecting a task the AMS-GUI loads a preset that adjusts input fields needed for this particular task. You can easily see what fields have been updated by a preset: they are colored green.
Check the GUI presets documentation for more information about presets.
XC functional¶
An important input option is the XC functional to use.
For this tutorial the default functional during the SCF is sufficient. So just leave this at the default value.
Tip
You should select a good XC functional and basis set to get accurate results. Numerical quality will also play a role. There often is a trade-off between speed and accuracy. Different job types could have different recommendations. E.g. for geometry optimizations, GGAs with dispersion correction and a DZP basis may be good enough, while for NMR you may want to use a hybrid functional with a TZ2P basis.
Basis set¶
With the Basis Set pull-down menu you select the basis set you want to use.
The menu gives access to the Basis Sets regularly used.
For this tutorial we will choose a very small basis set. This will yield less accurate results, but the calculation runs much faster. Obviously, if you want more accurate results you should use a better quality basis set. Thus:
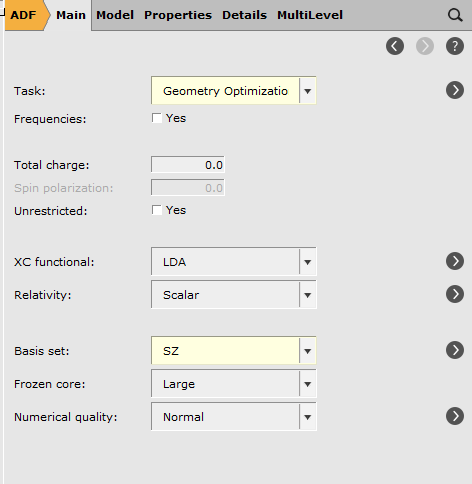
Tip
Colors of input field: changed by preset (green), by user (yellow), by both (red)
Numerical quality¶
The ADF engine uses a numerical integration scheme for virtually everything it may calculate. A numerical integration scheme generates a grid and corresponding weights. The default integration method in ADF and BAND is the Becke grid.
Another technical detail is that a density fitting method is used, for computational efficiency. The spline Zlm fit is the default fit method (the old Slater type fit is still available).
With the Numerical quality option you can select the quality of both the Becke integration and the spline Zlm fit at the same time.
Increasing the quality makes the results more accurate, but will require substantially more computation time.
Similarly, decreasing the quality will result in less accurate results, but you may get results faster.
The default value will in most cases be fine, certainly for this tutorial. If you go to the Details section you can set details of the integration scheme and fit method. However, the Numerical quality option in the main panel is the most convenient way to select the quality.
Geometry Convergence¶
 button next to the Task
button next to the Task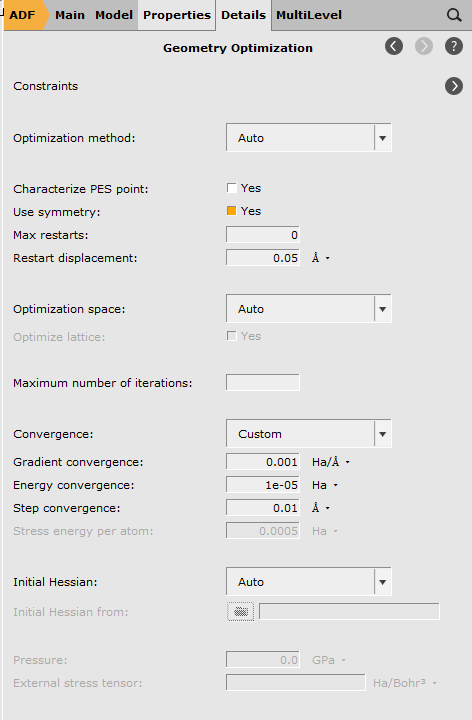
In this panel you can see the details of the geometry optimization, such as the maximum number of iterations and the convergence thresholds.
Tip
Click on a unit to change the unit, your choice will be remembered.
Use the GUI Preferences to reset all units to the default values.
As the current set up is fine, go back to the main panel:
Other input options¶
The panels on the right side contain many more input options. You select a panel with the menus in the panel bar, or by searching for a particular option. When searching for an option, any text in the panels will match, as well as from the help balloons. Also the corresponding AMS input keys will match.
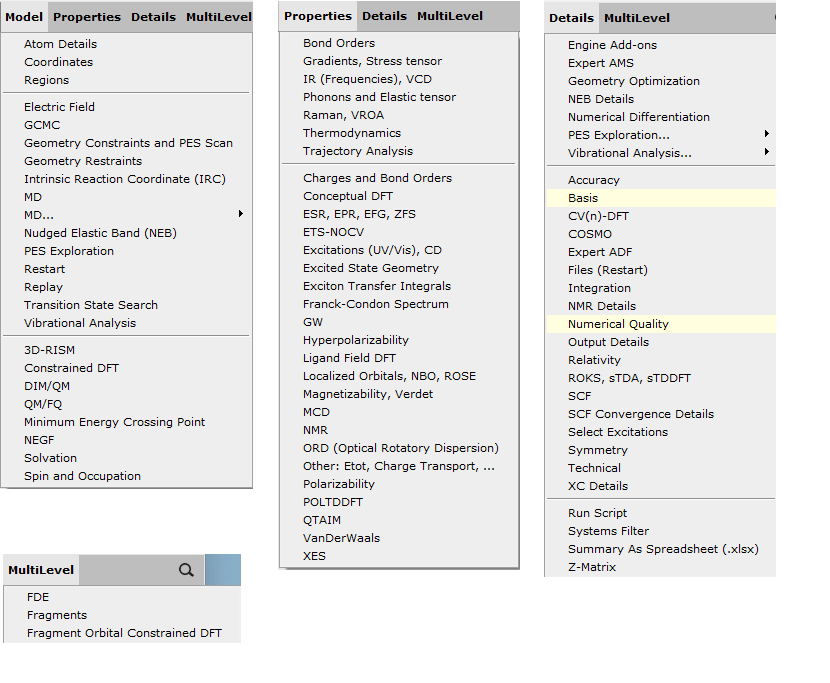
The menu items use a color coding to show you which panels have been affected by a preset (green), by the user (yellow), or both (red).
As we will not do anything special right now, you do not need to change anything in other panels.
Step 4: Run your calculation¶
Save your input and create a job script¶
Finally you will want to save your input.
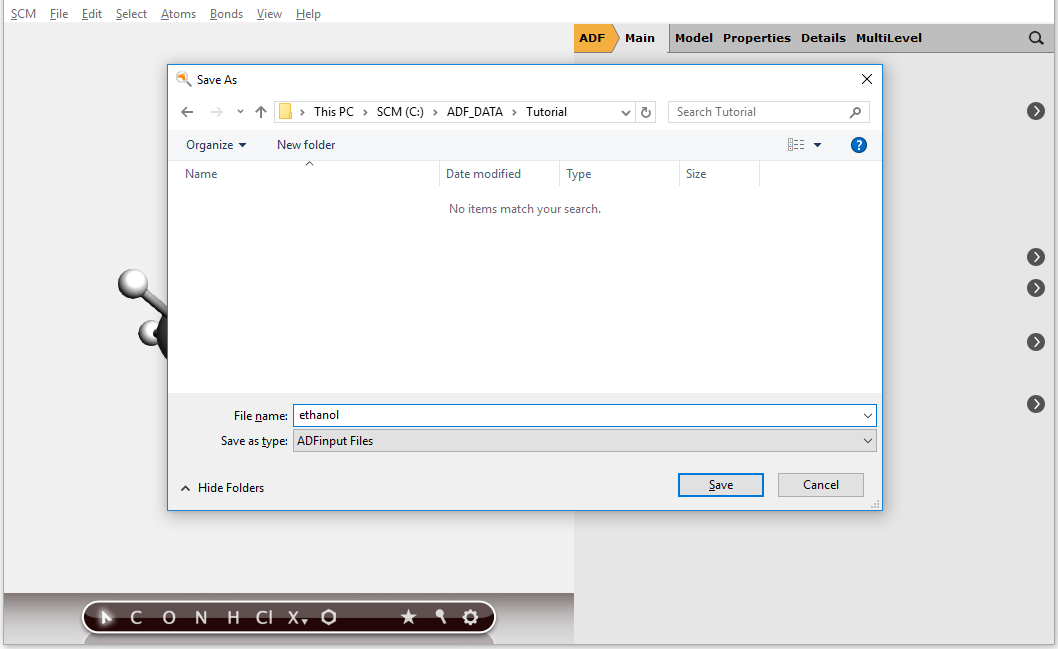
Now you have saved your current options and molecule information. The file will automatically get the extension ‘.ams’.
AMSinput has also created a corresponding script file. This script file has the same name, but with an extension ‘.run’ instead of ‘.ams’.
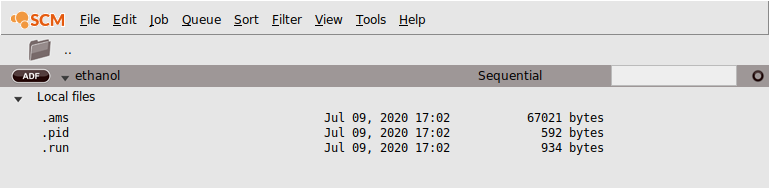
In the AMSjobs module you can see what files have been created:
You will see the .ams and .run files, and a .pid file that AMSjobs uses to store information. You might also see the picture that you saved, if you used the name ‘ethanol’ for it. Only the extensions are listed, so the real filenames are ethanol.ams, ethanol.run and ethanol.pid. Notice the job status icon (the open circle on the right) that AMSjobs uses to indicate a new job.
Run your calculation¶
To actually perform the calculation (the geometry optimization of the ethanol molecule), use the Job → Run menu command in AMSjobs:
This will execute the run script that has just been created. If you have never made changes in the AMSjobs setup, the default behavior is to run the job in the background on your local computer, using the Sequential queue. This queue will make sure that if you try to run more then one job at the same time, they will be run one after another.
Once your job starts running, AMSjobs will show the progress of the calculation: the last few lines of the logfile:
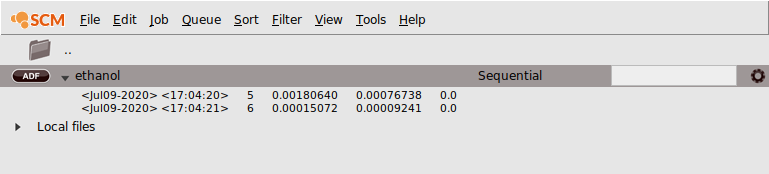
Note that while running, the job status symbol in AMSjobs changes.
If you wish to see the full logfile while the calculation is running, just click on the logfile lines displayed in the AMSjobs window:
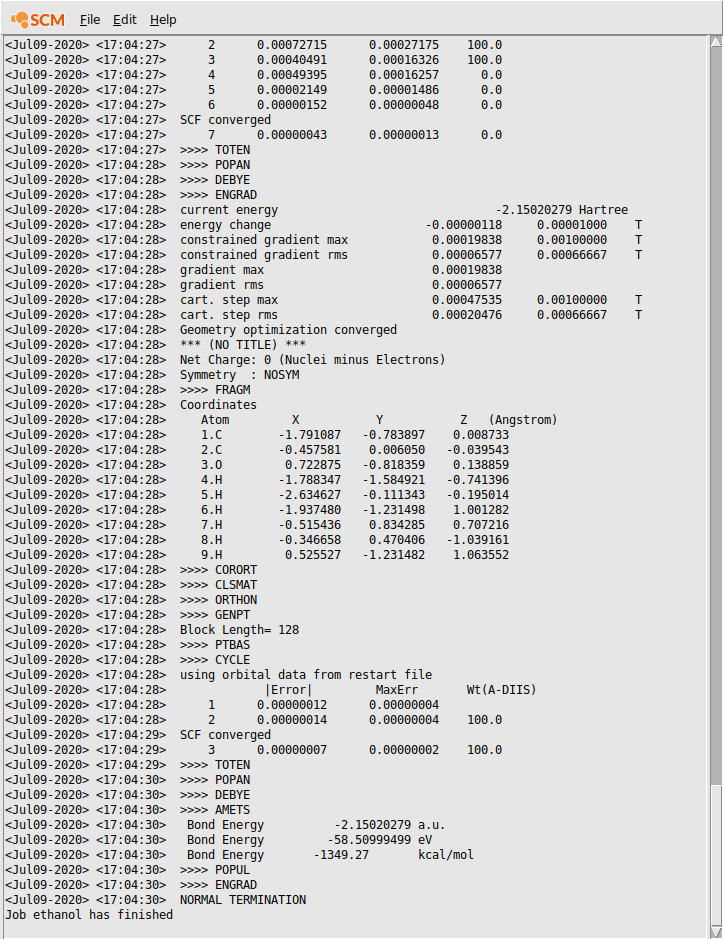
Now the logfile is showing in the AMStail window:
Step 5: Results of your calculation¶
Logfile: AMStail¶
The logfile is saved and extended by AMS as it is running. Normally it is most convenient to view it only in the AMSjobs window to prevent screen clutter.
Right now it is already showing in the AMStail window, and in the AMSjobs window, but you could have used any text editor.
Wait for the calculation to finish:
Close the AMStail window by using the File → Close menu command:
In the AMSjobs window, note that the job status icon has changed to indicate that the job is ready:

Files¶
AMS has created a couple of data files and a couple of text files. You can check this in the AMSjobs window:
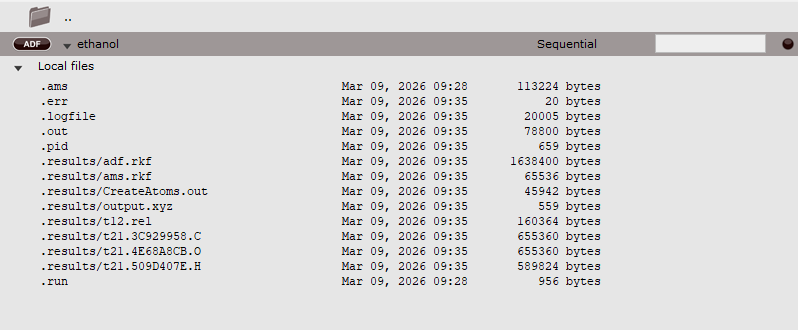
Note that the Local files are visible because earlier in the tutorial you already clicked on the triangle opening the details for this job.
The .ams file is the input as saved by AMSinput.
The .run file is the run script, also saved by AMSinput.
Tip
To change the .run file, if necessary, use the ‘Run Script’ panel in AMSinput (in the Details section). Your changes will be saved in the .ams file. If you make changes to the .run file itself they will be overwritten next time you save the job with AMSinput.
The .pid file is a file that contains your job status and configuration. It is used by AMSjobs.
The other files are produced by AMS. The .err file contains error messages, the .logfile shows the progress of the calculation, the .out file is the main (text) output file, and the other files are binary files that store results of the calculation.
Geometry changes: AMSmovie¶
AMS has optimized the geometry, and we can use the AMSmovie module to visualize the progress of the optimization. So let’s start AMSmovie using the SCM menu in your AMSinput window:
Tip
Press the space bar to start/stop playing in AMSmovie
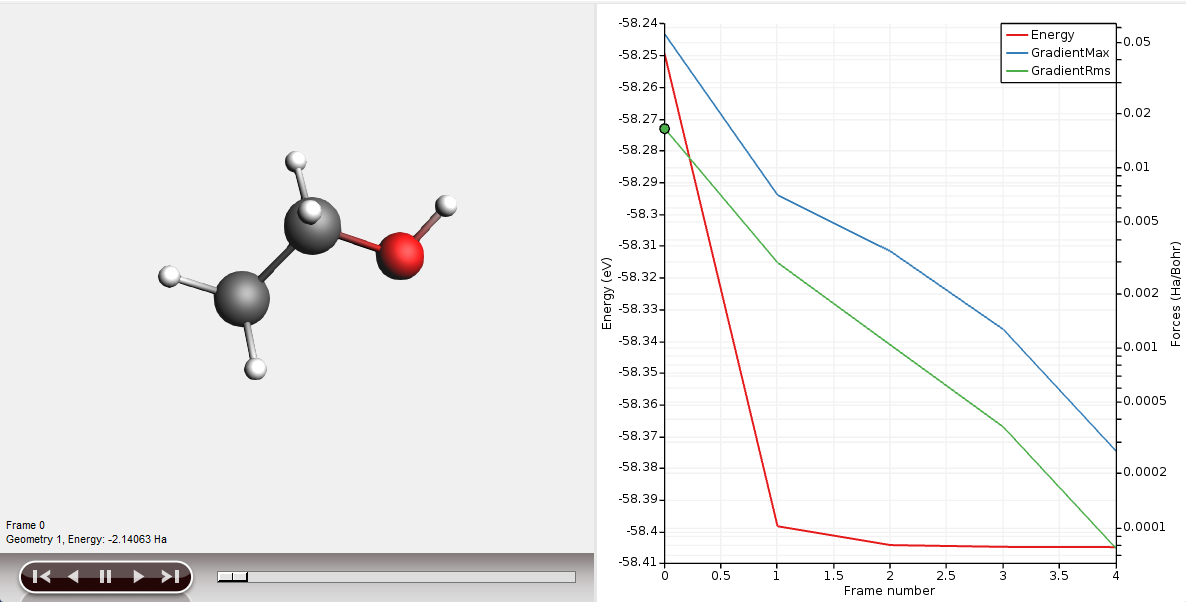
The AMSmovie module will display a movie of the geometry optimization.
Tip
Use the slider to quickly move through the frames.
Use the left and right arrow keys to single step through the frames.
You control playing with the buttons. When your mouse pointer is above any of the buttons, and not moving, a balloon, or tooltip, will pop up showing what that particular button will do.
The graph on the right-hand side shows the energy and forces as function of the geometry step.
You can show several graphs for different properties at the same time:
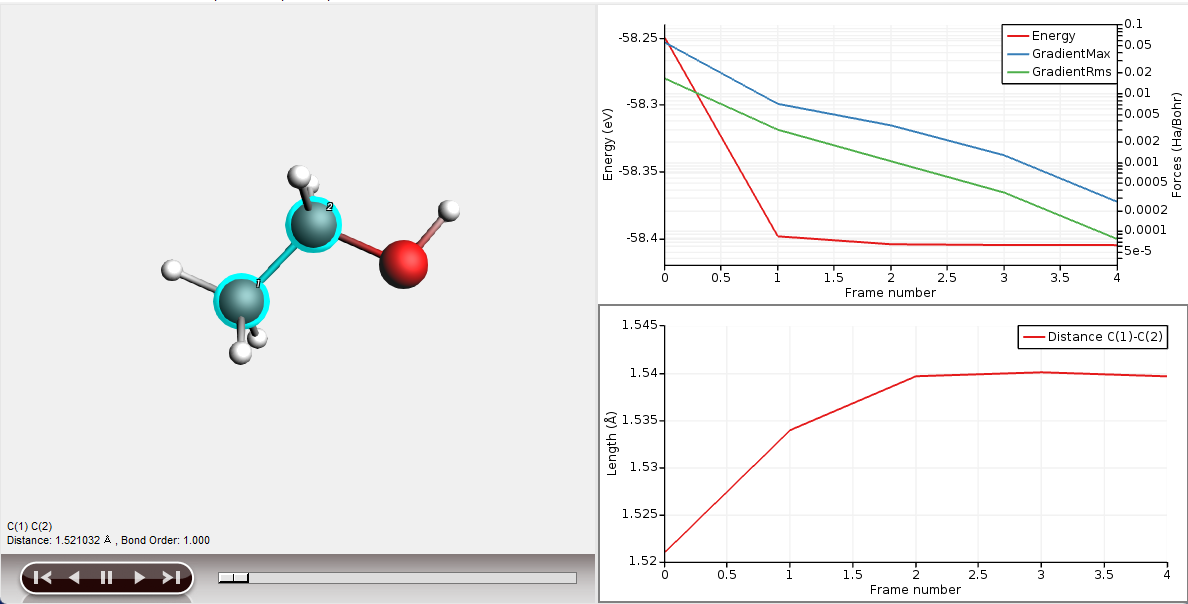
Now you have two graphs. One of them is the ‘active’ graph. When you make a new graph it will always be the active graph. You can also make a graph active by clicking on it.
When you select a property from the Graph menu (Energy, Distance and so on) that property will be plotted in the active graph.
You can also have multiple curves in one graph, if possible: one property per Y-axis. You may have several curves on the same Y axes if they are using the same unit (all Angstroms for example):
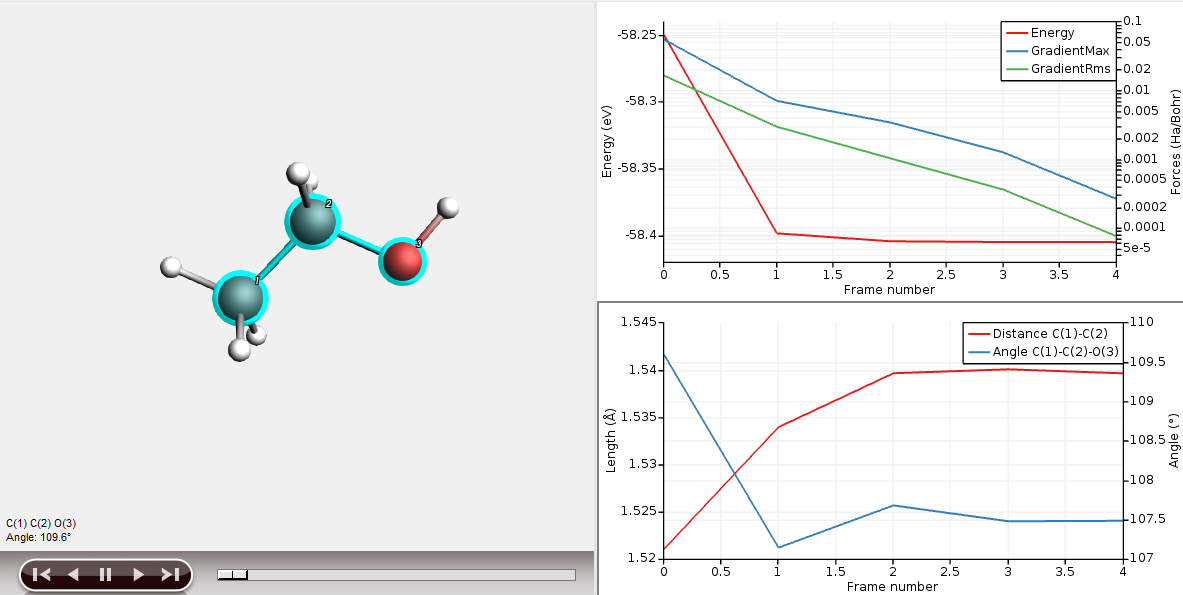
Another feature is that you can click on a point in one of the graphs. It will be marked, the movie will jump to that particular step, and if you have more then one graph the corresponding point(s) will also be marked in the other graphs.
To rotate, translate, or zoom the picture, use your mouse, just as in AMSinput.
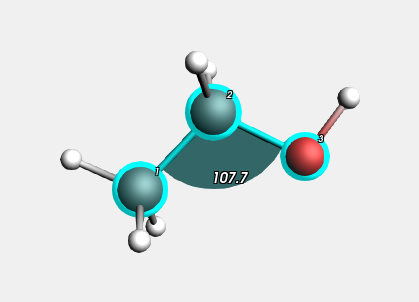
Selecting atoms provides information about atoms, bonds, etc. in the information field below the molecule editor pane. The information will be updated when you go to another point in the movie (a different geometry). You can see examples of these in the pictures above.
You can also show this information in the 3D window:
The angle will be visually added to your molecule :
Orbital energy levels: AMSlevels¶
Tip
In AMSjobs right-click on a job name to go quickly select it and show the SCM menu
AMSlevels will start and show a diagram of the energy levels of the ethanol molecule.
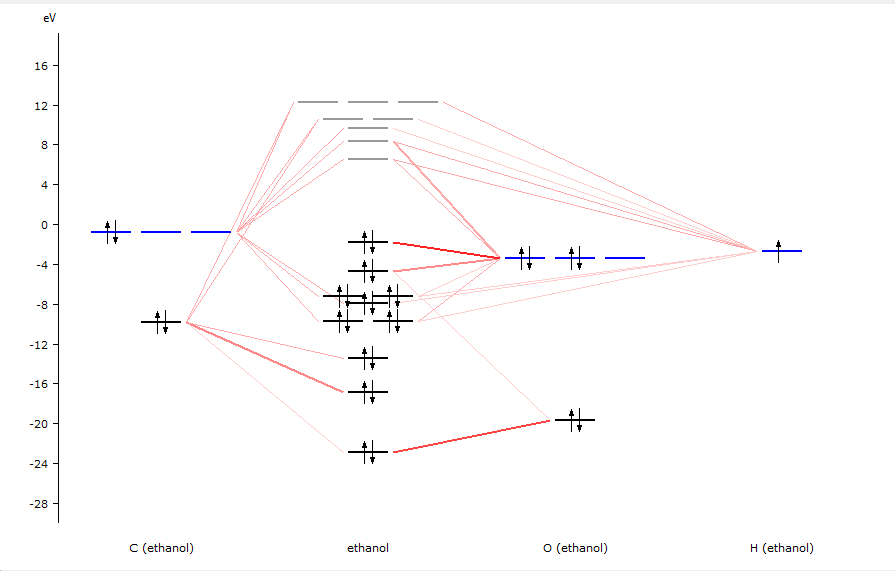
In the diagram you can see from what fragment types the molecular levels are composed.
Balloons will pop up with information about the level at the mouse position: The MO number, eigenvalue, occupation, and how it is composed of SFOs (symmetrized fragment orbitals).
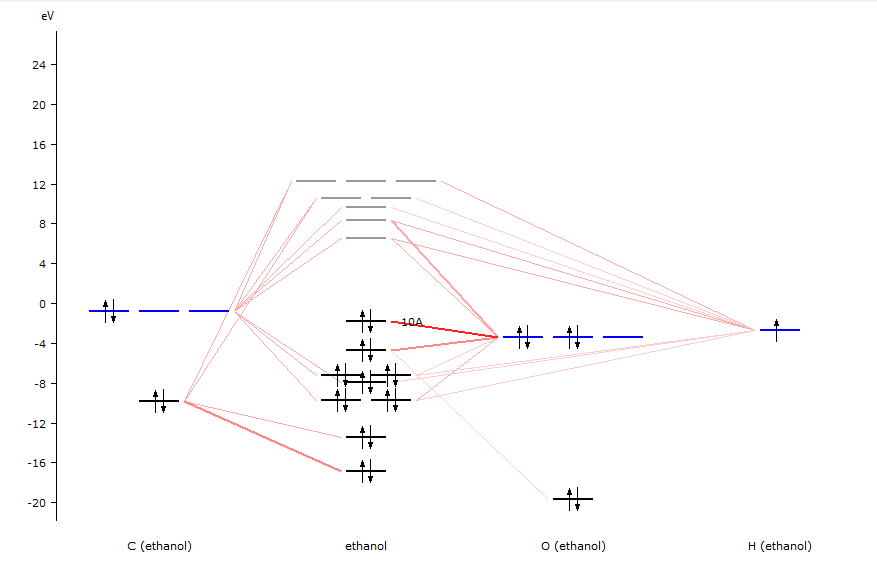
The labels of the orbitals may be different as they depend on the symmetry of your molecule.
To actually see the orbital, select the orbital from the top of the pop-up menu:
A window with a picture of the orbital should appear.
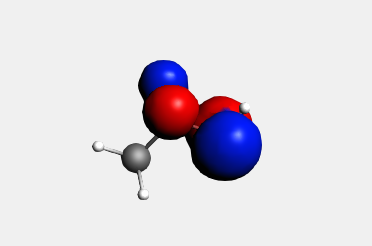
You can move (rotate, translate and zoom) the orbital with your mouse.
Electron density, potential and orbitals: AMSview¶
AMSview will start up and show a picture of your molecule and a panel where calculated properties will be listed:
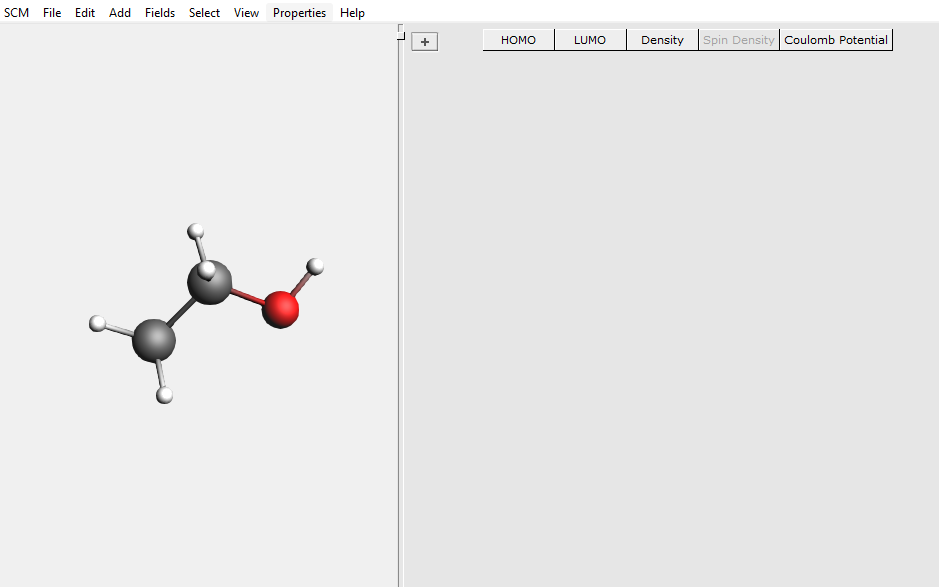
You can use AMSview to visualize all kinds of ‘field’ related properties: densities, orbitals, potentials, etc. You actually have already used it before: the picture of the orbital that was created using AMSlevels was shown by AMSview.
Use the mouse to rotate, translate or zoom, as in AMSinput.
In the Properties menu there are some pre-defined things to visualize: density, spin-density, HOMO, LUMO and more. If you select one of these, you will see the corresponding item immediately. However, AMSview can do much more and gives you lots of control.
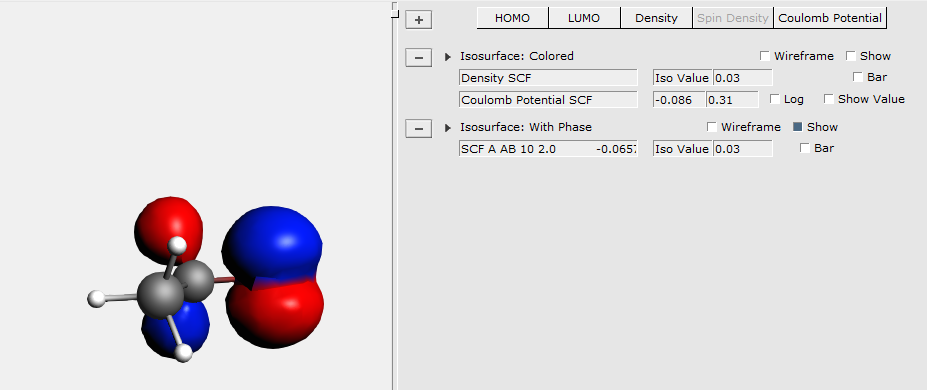
For example, lets show a density isosurface, colored by the electrostatic potential:
At the right side extra controls will be created. AMSview creates such controls for all visual items and special fields (surfaces, planes, calculated fields, etc.) that you add.
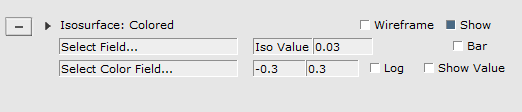
In this particular case the controls contains two pull-down menus that you use to select the fields that you want to visualize.
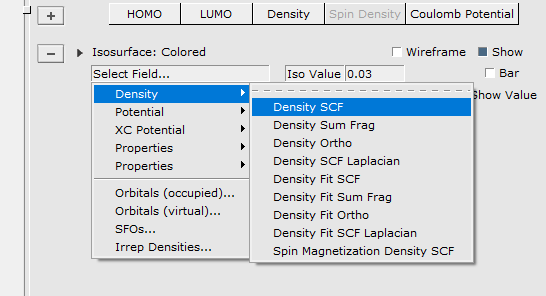

To demonstrate some other possibilities of AMSview, do the following:
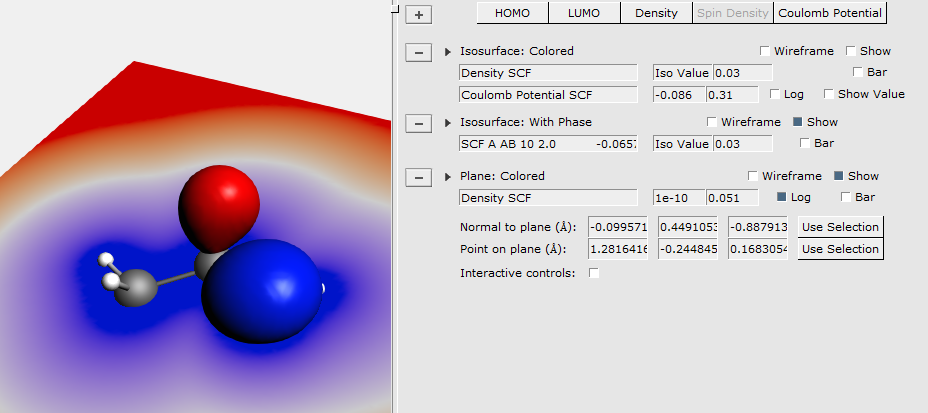
You can save the picture you create using the Save Picture menu command:
A picture with the (file)name you specified has been created.
You might want to explore some more of the possibilities of AMSview on your own. Many different properties can be visualized as you probably have noticed in the pull-down menus.
Browsing the Output: AMSoutput¶
The output file (tutorial.out) is a plain text file. You can view it with your favorite text editor (or text viewer). You can also use the AMSoutput GUI module which provides a convenient way to check the results:
The AMSoutput program will start showing the results of your calculation, and via the menu you jumped to the section with bonding energy decomposition details:
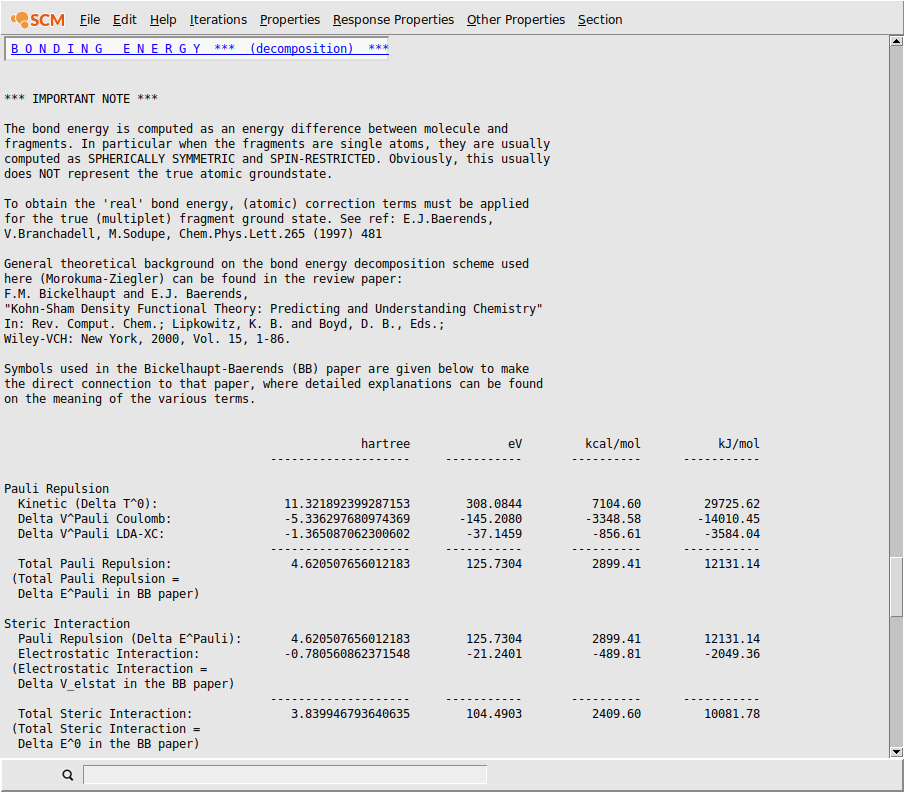
You can use the menus to go to different parts of the output file, or you can just use the scroll bar. If a menu option is shaded, this means that no corresponding section of the output is available.
Tip
Click text highlighted in blue to jump to the next section with the same title, if present.
Tip
Use the search box at the bottom of the AMSoutput window (cmd/ctrl-F)
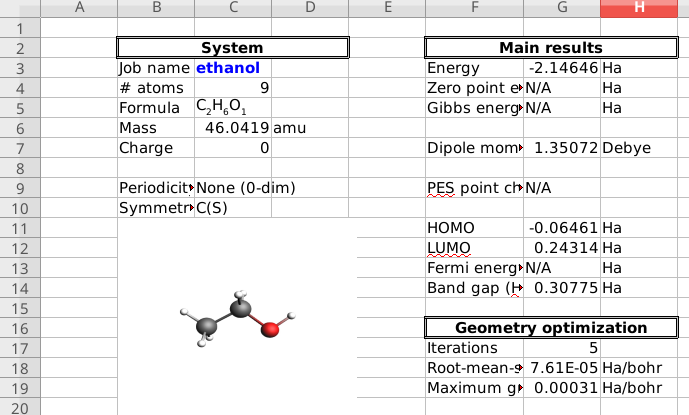
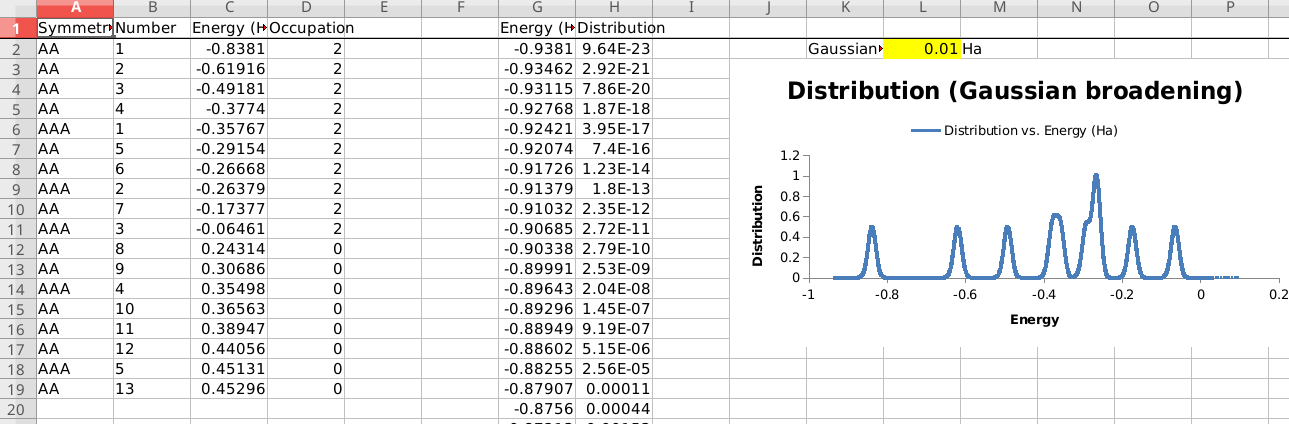
Convert results to spreadsheet (.xlsx)¶
You can also export the results of a finished calculation to spreadsheet (.xlsx) format, that you can open in e.g., Microsoft Excel or LibreOffice Calc.
Only the most common types of results are exported to the spreadsheet. For example, general information about the system, the orbital energies, and the atomic positions and atomic charges. For a full list, the spreadsheets documentation.
Close all open GUI windows¶
As we are now done with tutorial 1, close all windows that belong to this tutorial:
All open windows from the AMS-GUI will be closed.