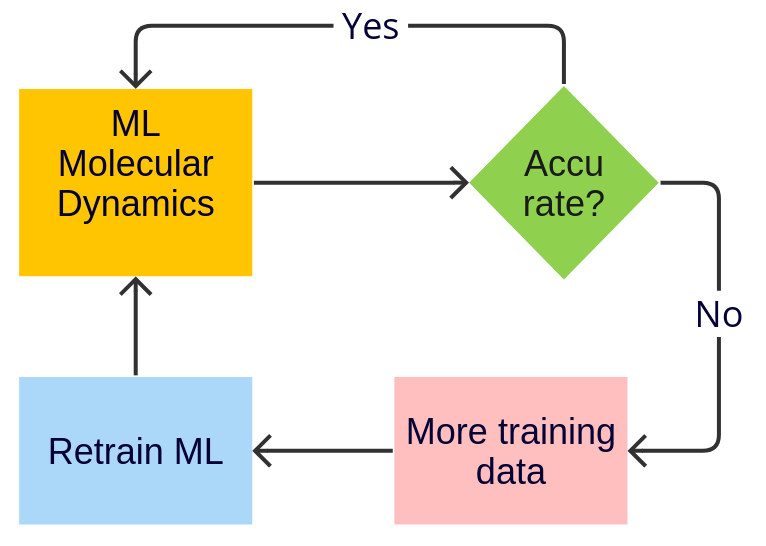
Workflows and Automation¶
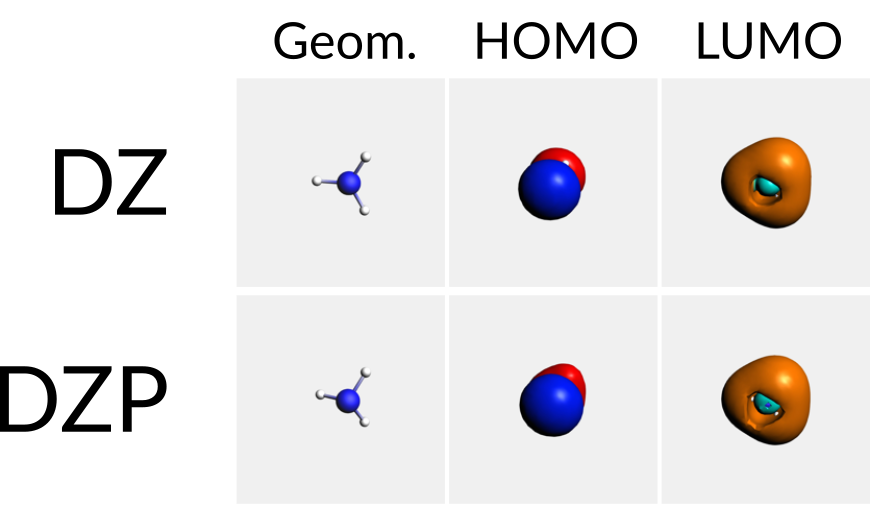
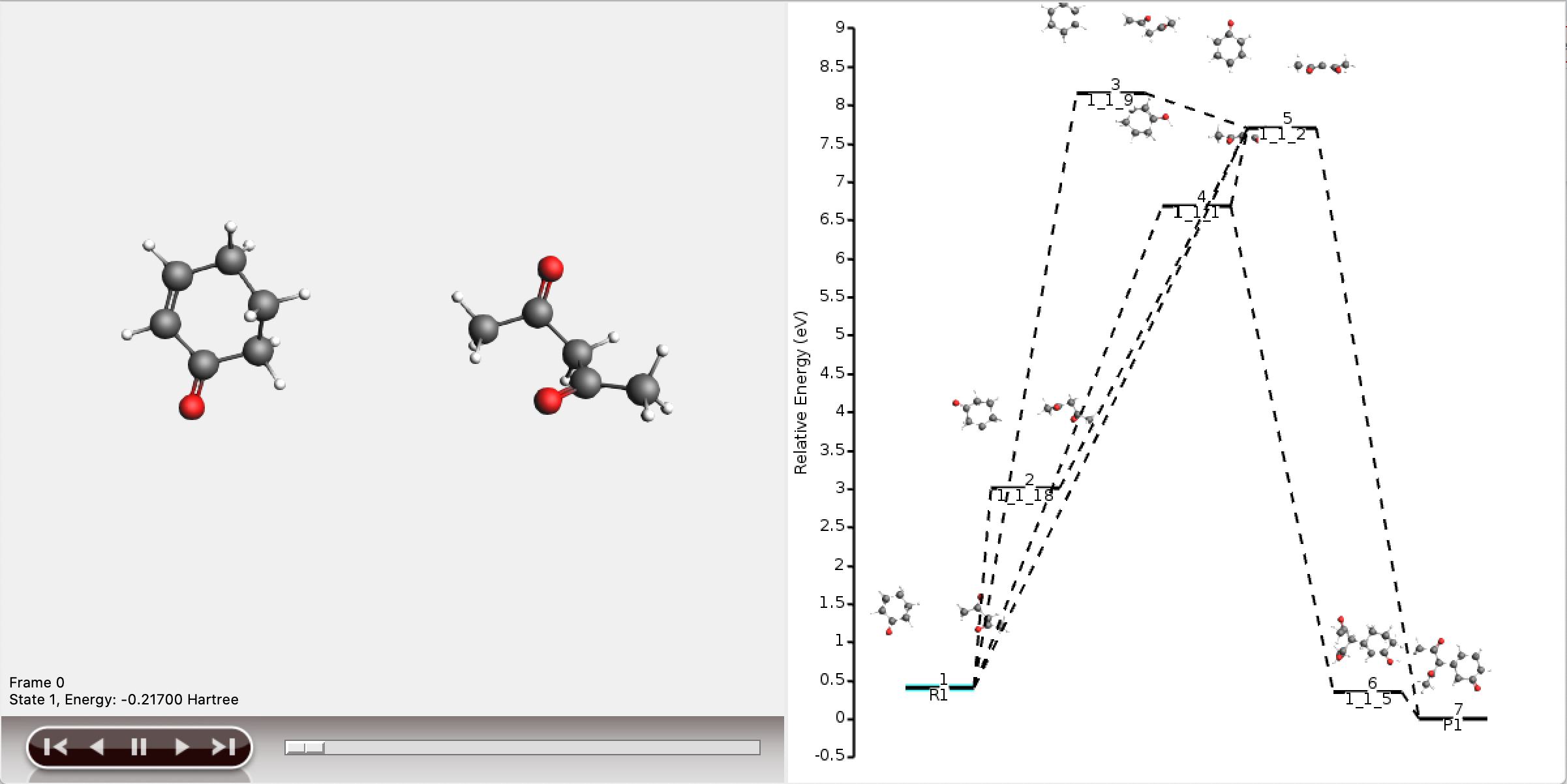
Generating a batch of jobs and collecting results: Basis Set Effects for NH3 Geometry
Keywords: batch jobs from the GUI, AMSjobs, AMSprep and AMSreport, basis sets
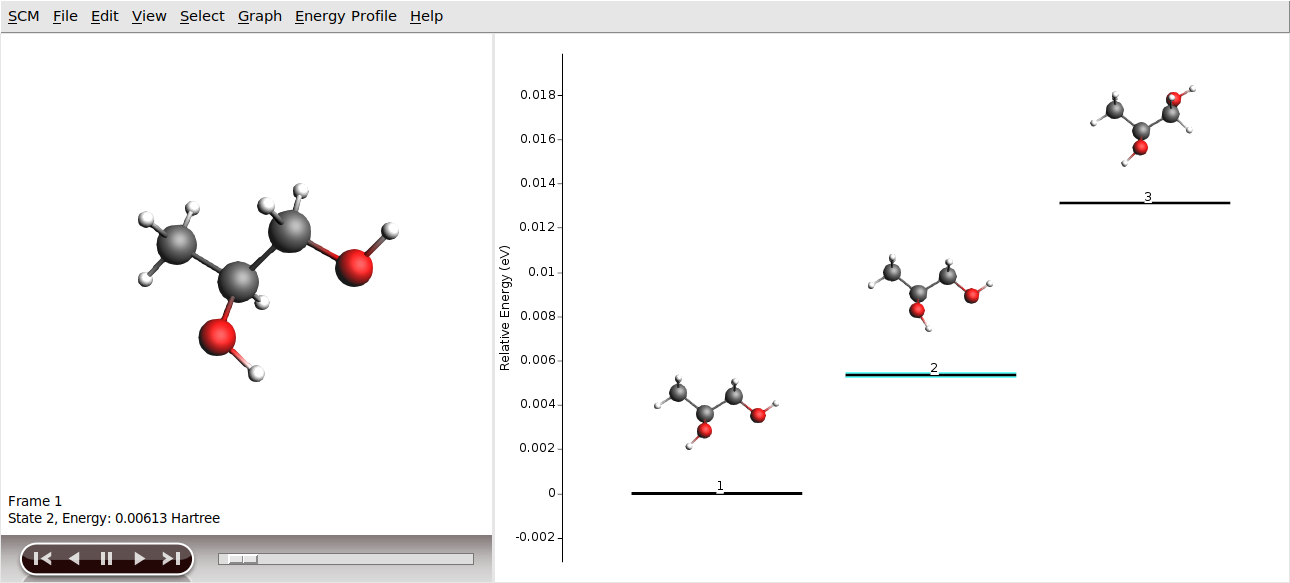
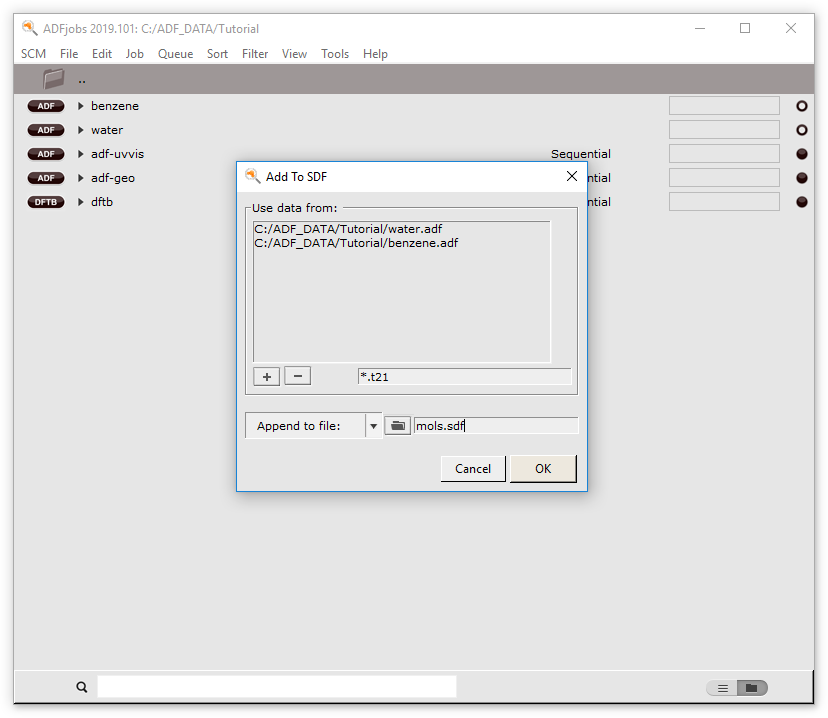
Multiple molecules multiple methods
Keywords: GUI workflows, SDF files, multiple molecules in AMSinput

Multiscale modeling of OLED devices
Keywords: organic electronics, OLED, physical vapor deposition, thin-film morphologies, Ionization Potentials (IP), Electron Affinities (EA)