Phase Transitions in the ZGB Model¶
Note
To follow this tutorial, either:
Download
PhaseTransitions.py(run as$AMSBIN/amspython PhaseTransitions.py).Download
PhaseTransitions.ipynb(see also: how to install Jupyterlab)
This example is inspired in the seminal paper: Kinetic Phase Transitions in an Irreversible Surface-Reaction Model by Robert M. Ziff, Erdagon Gulari, and Yoav Barshad in 1986 Phys. Rev. Lett. 56, (1986) 2553. The authors proposed a simple model for catalytic reactions of carbon monoxide oxidation to carbon dioxide on a surface. This model is now known as the Ziff-Gulari-Barshad (ZGB) model after their names. While the model leaves out many important steps of the real system, it exhibits interesting steady-state off-equilibrium behavior and two types of phase transitions, which actually occur in real systems. Please refer to the original paper for more details. In this example, we will analyze the effect of changing the composition of the gas phase, namely partial pressures for \(O_2\) and \(CO\), in the \(CO_2\) Turnover frequency (TOF) in the ZGB model.
The first step is to import all packages we need:
import multiprocessing
import numpy
import scm.plams
import scm.pyzacros as pz
First must define the system. So, we have to specify species, lattice, cluster expansion, and mechanisms. As we said above, we will use the ZGB model, which consists on:
1. Three gas species: \(CO\), \(O_2\), and \(CO_2\).
Notice that the gas_energy by default is zero unless otherwise
stated. That’s the case for \(CO\) and \(O2\), which are used as
energy references.
CO_gas = pz.Species('CO')
O2_gas = pz.Species('O2')
CO2_gas = pz.Species('CO2", gas_energy=-2.337)
2. Three surface species: \(*\), \(CO^*\), \(O^*\). The species \(*\) represents the empty adsorption site. All of them have denticity equal to 1.
s0 = pz.Species("*", 1)
CO_ads = pz.Species("CO*", 1)
O_ads = pz.Species("O*", 1)
3. A rectangular lattice with a single site type.
lattice = pz.Lattice(lattice_type=pz.Lattice.RECTANGULAR, lattice_constant=1.0, repeat_cell=[50, 50])
4. Two clusters in the cluster-expansion Hamiltonian: \(CO^*\)-bs and \(O^*\)-bs. They are attached to a single binding site. No lateral interactions are considered.
CO_point = pz.Cluster(species=[CO_ads], energy=-1.3)
O_point = pz.Cluster(species=[O_ads], energy=-2.3)
cluster_expansion = [CO_point, O_point]
5. Three irreversible events: adsorption of \(CO\), dissociative adsorption of \(O_2\), and \(CO\) oxidation.
# CO_adsorption:
CO_adsorption = pz.ElementaryReaction(
initial=[s0, CO_gas], final=[CO_ads], reversible=False, pre_expon=10.0, activation_energy=0.0
)
# O2_adsorption:
O2_adsorption = pz.ElementaryReaction(
initial=[s0, s0, O2_gas],
final=[O_ads, O_ads],
neighboring=[(0, 1)],
reversible=False,
pre_expon=2.5,
activation_energy=0.0,
)
# CO_oxidation:
CO_oxidation = pz.ElementaryReaction(
initial=[CO_ads, O_ads],
final=[s0, s0, CO2_gas],
neighboring=[(0, 1)],
reversible=False,
pre_expon=1.0e20,
activation_energy=0.0,
)
mechanism = [CO_adsorption, O2_adsorption, CO_oxidation]
Now, we initialize the pyZacros environment.
scm.pyzacros.init()
PLAMS working folder: /home/user/pyzacros/examples/ZiffGulariBarshad/plams_workdir
This calculation is relatively fast. On a typical laptop, it should take
around 1 min to complete. However, to illustrate how to run several in
parallel Zacros calculations, we’ll use the plams.JobRunner class,
which easily allows us to run as many parallel instances as we request.
In this case, we choose to use the maximum number of simultaneous
processes (maxjobs) equal to the number of processors in the
machine. Additionally, by setting nproc = 1 we establish that only
one processor will be used for each zacros instance.
maxjobs = multiprocessing.cpu_count()
scm.plams.config.default_jobrunner = scm.plams.JobRunner(parallel=True, maxjobs=maxjobs)
scm.plams.config.job.runscript.nproc = 1
print("Running up to {} jobs in parallel simultaneously".format(maxjobs))
Running up to 8 jobs in parallel simultaneously
Now we have to set up the calculation using a Settings object.
Firstly, we set a reactants gas phase composition of 45% \(CO\) and
55% \(O_2\). Notice that we assumed that the gas phase is composed
only of \(CO\) and \(O_2\); thus,
\(x_\text{CO} +x_{\text{O}_2} =1\). Keep in mind that these values
are actually not affecting the calculation because, later on, these are
the ones we will modify systematically. Then, we set the physical
parameters as temperature (500 K) and pressure (1 bar). Finally, we set
the maximum time for the simulation to 10 s (max_time), and we save
snapshots of the lattice state every 0.5 s (snapshots) and the
number of species every 0.1 s (species_numbers). The last line sets
the random seed to make the calculations reproducible.
sett = pz.Settings()
sett.molar_fraction.CO = 0.45
sett.molar_fraction.O2 = 0.55
sett.temperature = 500.0
sett.pressure = 1.0
sett.max_time = 10.0
sett.snapshots = ("time", 0.5)
sett.species_numbers = ("time", 0.1)
sett.random_seed = 953129
The calculation parameters setup is ready. Therefore, we can proceed to
run the calculations. In this instance, we are interested in exploring
the production of \(CO_2\) by ranging the \(CO\) molar fraction
x_CO from 0.02 to 0.8 in steps of 0.01. Remember that \(O_2\)
molar fraction is set to satisfy \(x_\text{CO}+x_{\text{O}_2}=1\).
The loop creates one ZacrosJob for each value of x_CO
(condition) using the settings and system’s properties defined above and
executing it by invoking the function run(). Notice that results are
stored in the vector results for further analysis once they are
finished. In the output, observe that pyZacros creates a new folder
for each condition, following the sequence plamsjob.002,
plamsjob.003, plamsjob.004, and so on for
x_CO=0.20, 0.21, 0.22, ... respectively. The second loop calls the
method job.ok() of every job to ensure that every calculation was
completed successfully and wait for all parallel processes to complete
before proceeding to access the results.
x_CO = numpy.arange(0.2, 0.8, 0.01)
results = []
for x in x_CO:
sett.molar_fraction.CO = x
sett.molar_fraction.O2 = 1.0 - x
job = pz.ZacrosJob(settings=sett, lattice=lattice, mechanism=mechanism, cluster_expansion=cluster_expansion)
results.append(job.run())
for i, x in enumerate(x_CO):
if not results[i].job.ok():
print("Something went wrong with condition xCO={}!".format(x))
[02.02|22:23:20] JOB plamsjob STARTED
[02.02|22:23:20] JOB plamsjob STARTED
[02.02|22:23:20] Renaming job plamsjob to plamsjob.002
[02.02|22:23:20] JOB plamsjob STARTED
[02.02|22:23:20] Renaming job plamsjob to plamsjob.003
[02.02|22:23:20] JOB plamsjob STARTED
[02.02|22:23:20] Renaming job plamsjob to plamsjob.004
[02.02|22:23:20] JOB plamsjob STARTED
[02.02|22:23:20] Renaming job plamsjob to plamsjob.005
[02.02|22:23:20] JOB plamsjob STARTED
...
[02.02|22:23:28] JOB plamsjob.054 SUCCESSFUL
[02.02|22:23:28] JOB plamsjob.056 SUCCESSFUL
[02.02|22:23:28] JOB plamsjob.061 FINISHED
[02.02|22:23:28] JOB plamsjob.055 SUCCESSFUL
[02.02|22:23:28] JOB plamsjob.060 SUCCESSFUL
[02.02|22:23:28] JOB plamsjob.058 SUCCESSFUL
[02.02|22:23:28] Waiting for job plamsjob.058 to finish
[02.02|22:23:28] JOB plamsjob.059 SUCCESSFUL
[02.02|22:23:28] Waiting for job plamsjob.061 to finish
[02.02|22:23:28] JOB plamsjob.061 SUCCESSFUL
If the script worked successfully, you should have seen several
SUCCESSFUL messages at the output’s end.
Now we need to extract the results we want, in this case, the average
coverage and the turnover frequency of \(CO_2\), and store them
conveniently, as in arrays. We use the turnover frequency method for
the latter, and average coverage for the former, specifying that we want
to use the last five lattice states (last=5):
ac_O = []
ac_CO = []
TOF_CO2 = []
for i, x in enumerate(x_CO):
ac = results[i].average_coverage(last=5)
TOFs, _, _, _ = results[i].turnover_frequency()
ac_O.append(ac["O*"])
ac_CO.append(ac["CO*"])
TOF_CO2.append(TOFs["CO2"])
Finally, we just nicely print the results in a table.
print("----------------------------------------------")
print("%4s" % "cond", "%8s" % "x_CO", "%10s" % "ac_O", "%10s" % "ac_CO", "%10s" % "TOF_CO2")
print("----------------------------------------------")
for i, x in enumerate(x_CO):
print("%4d" % i, "%8.2f" % x_CO[i], "%10.6f" % ac_O[i], "%10.6f" % ac_CO[i], "%10.6f" % TOF_CO2[i])
----------------------------------------------
cond x_CO ac_O ac_CO TOF_CO2
----------------------------------------------
0 0.20 0.998000 0.000000 0.049895
1 0.21 1.000000 0.000000 0.046695
2 0.22 1.000000 0.000000 0.051747
3 0.23 0.998880 0.000000 0.053747
4 0.24 0.997600 0.000000 0.061937
5 0.25 0.996560 0.000000 0.087368
6 0.26 0.998960 0.000000 0.073600
7 0.27 0.998160 0.000000 0.085537
8 0.28 0.997680 0.000000 0.098905
9 0.29 0.994560 0.000000 0.111011
10 0.30 0.995840 0.000000 0.123811
11 0.31 0.996320 0.000000 0.134463
12 0.32 0.991760 0.000000 0.163811
13 0.33 0.991680 0.000000 0.165958
14 0.34 0.989360 0.000000 0.224589
15 0.35 0.981680 0.000000 0.273811
16 0.36 0.962960 0.000240 0.319789
17 0.37 0.963440 0.000160 0.352800
18 0.38 0.938000 0.000320 0.404000
19 0.39 0.936640 0.000400 0.508589
20 0.40 0.898320 0.000480 0.577095
21 0.41 0.872800 0.000880 0.640084
22 0.42 0.868560 0.001280 0.756400
23 0.43 0.827680 0.001680 0.895537
24 0.44 0.808400 0.002160 1.002884
25 0.45 0.759280 0.002960 1.266484
26 0.46 0.732080 0.006480 1.337684
27 0.47 0.657520 0.009200 1.523621
28 0.48 0.641520 0.010880 1.729979
29 0.49 0.592960 0.020000 1.872905
30 0.50 0.567280 0.022480 2.108442
31 0.51 0.567920 0.024880 2.259958
32 0.52 0.432000 0.066880 2.537432
33 0.53 0.402960 0.078960 2.770863
34 0.54 0.062480 0.757680 2.093579
35 0.55 0.019520 0.913760 1.770484
36 0.56 0.000000 1.000000 0.940568
37 0.57 0.000000 1.000000 0.489347
38 0.58 0.000000 1.000000 0.356063
39 0.59 0.000000 1.000000 0.253705
40 0.60 0.000000 1.000000 0.221453
41 0.61 0.000000 1.000000 0.154316
42 0.62 0.000000 1.000000 0.107621
43 0.63 0.000000 1.000000 0.066926
44 0.64 0.000000 1.000000 0.059116
45 0.65 0.000000 1.000000 0.042695
46 0.66 0.000000 1.000000 0.040632
47 0.67 0.000000 1.000000 0.029074
48 0.68 0.000000 1.000000 0.012905
49 0.69 0.000000 1.000000 0.009411
50 0.70 0.000000 1.000000 0.008863
51 0.71 0.000000 1.000000 0.007221
52 0.72 0.000000 1.000000 0.003768
53 0.73 0.000000 1.000000 0.002063
54 0.74 0.000000 1.000000 0.002105
55 0.75 0.000000 1.000000 0.000611
56 0.76 0.000000 1.000000 0.000758
57 0.77 0.000000 1.000000 0.000400
58 0.78 0.000000 1.000000 0.000000
59 0.79 0.000000 1.000000 0.000758
60 0.80 0.000000 1.000000 0.000589
The above results are the final aim of the calculation. However, we can take advantage of python libraries to visualize them. Here, we use matplotlib. Please check the matplotlib documentation for more details at matplotlib. The following lines of code allow visualizing the effect of changing the \(CO\) molar fraction on the average coverage of \(O*\) and \(CO*\) and the production rate of \(CO_2\):
import matplotlib.pyplot as plt
fig = plt.figure()
ax = plt.axes()
ax.set_xlabel("Molar Fraction CO", fontsize=14)
ax.set_ylabel("Coverage Fraction (%)", color="blue", fontsize=14)
ax.plot(x_CO, ac_O, color="blue", linestyle="-.", lw=2, zorder=1)
ax.plot(x_CO, ac_CO, color="blue", linestyle="-", lw=2, zorder=2)
plt.text(0.3, 0.9, "O", fontsize=18, color="blue")
plt.text(0.7, 0.9, "CO", fontsize=18, color="blue")
ax2 = ax.twinx()
ax2.set_ylabel("TOF (mol/s/site)", color="red", fontsize=14)
ax2.plot(x_CO, TOF_CO2, color="red", lw=2, zorder=5)
plt.text(0.37, 1.5, "CO$_2$", fontsize=18, color="red")
plt.show()

This model assumes that gas-phase molecules of \(CO\) and \(O_2\) are adsorbed immediately on empty sites, and when the \(0*\) and \(CO*\) occupy adjacent sites, they react immediately. This model is intrinsically irreversible because the molecules are sticky to their original sites and remain stationary until they are removed by a reaction. This leads to the figure above having three regions:
Oxygen poisoned state, \(x_\text{CO}<0.32\).
Reactive state \(0.32<x_\text{CO}<0.55\).
CO poisoned state \(x_\text{CO}>0.55\).
The first transition at \(x_\text{CO}=0.32\) is continuous, and therefore it is of the second order. The second transition at \(x_\text{CO}=0.55\) occurs abruptly, implying that this is of a first-order transition. As you increase the simulation time, the transition becomes more abrupt. We will discuss this effect in the next tutorial Ziff-Gulari-Barshad model: Steady State Conditions.
pyZacros also offers some predefined plot functions that use
matplotlib as well. For example, it is possible to see a typical
reactive state configuration \(x_\text{CO}=0.54\) and one in the
process of being poisoned by \(CO\) (\(x_\text{CO}=0.55\)). Just
get the last lattice state with the last_lattice_state() function
and visualize it with plot(). See the code and figures below.
The state at \(x_\text{CO}=0.54\) is a prototypical steady-state,
results[33].last_lattice_state().plot()
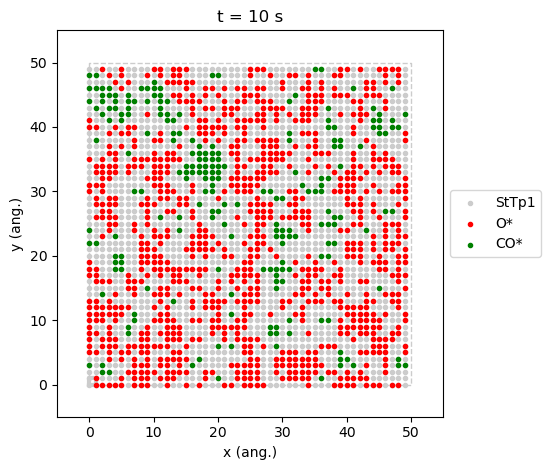
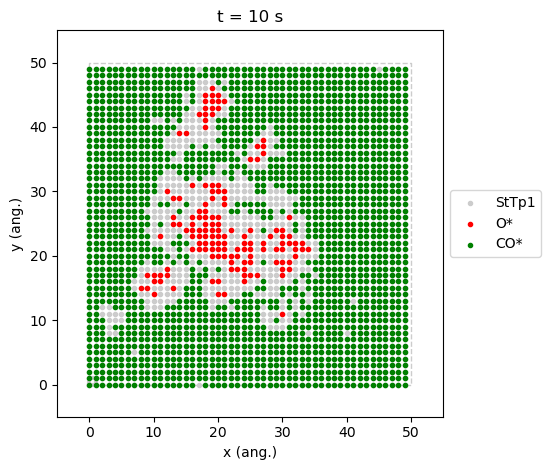
contrary to the one at \(x_\text{CO}=0.55\), is a good example where we can see the two phases coexisting.
results[34].last_lattice_state().plot()
In the previous paragraph, we introduced the concept of steady-state. However, let’s define it slightly more formally. For our study system, the steady-state for a given composition is characterized when the derivative of the \(CO_2\) production (TOF) with respect to time is zero and remains so:
pyZacros also offers the function plot_molecule_numbers() to
visualize the molecule numbers and its first derivative as a function of
time. See code and figures below:
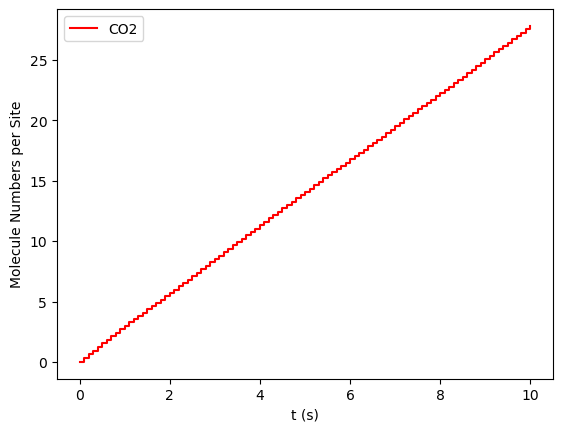
results[33].plot_molecule_numbers(["CO2"], normalize_per_site=True)
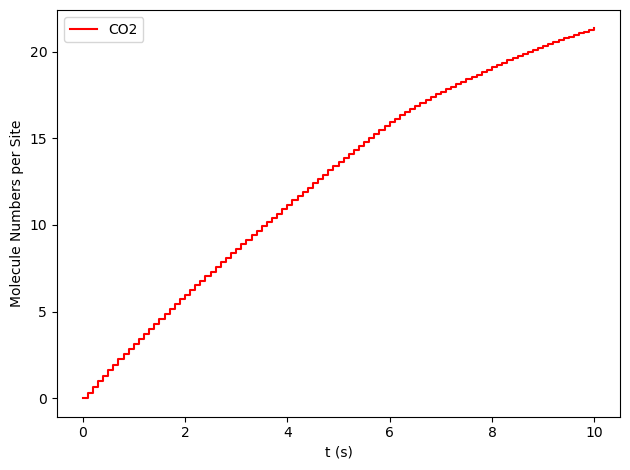
results[34].plot_molecule_numbers(["CO2"], normalize_per_site=True)
results[33].plot_molecule_numbers(["CO2"], normalize_per_site=True, derivative=True)
results[34].plot_molecule_numbers(["CO2"], normalize_per_site=True, derivative=True)
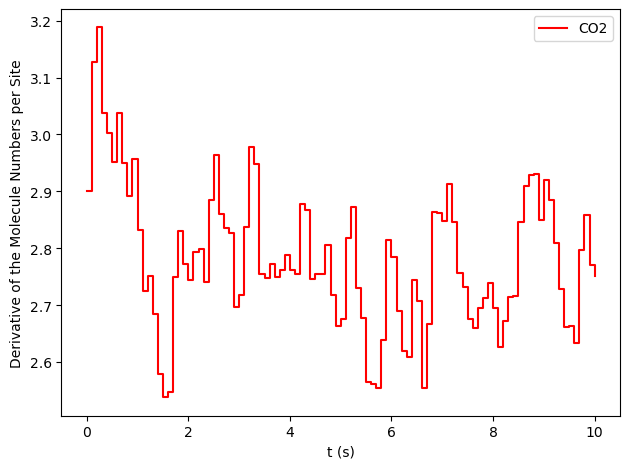
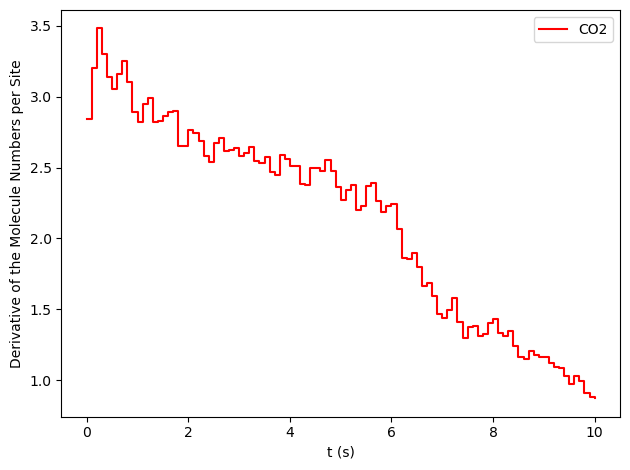
From the figures above, it is clear that we have reached a steady-state for \(x_\text{CO}=0.54\). Notice that the first derivative is approximately constant at 2.7 mol/s/site within a tolerance of 5 mol/s/site. Contrary, this is not the case of \(x_\text{CO}=0.55\), where the first derivative continuously decreases.
Now, we can close the pyZacros environment:
scm.pyzacros.finish()
[02.02|22:24:09] PLAMS run finished. Goodbye
Visualization and Post-processing¶
pyZacros allows you to load the results from previously completed calculations. This allows you to perform additional visualization or data analysis without having to redo the entire calculation. An example is provided below, reproducing some of the graphs discussed in this tutorial:
import scm.pyzacros as pz
# xCO=0.54
job = pz.ZacrosJob.load_external( path="plams_workdir/plamsjob.034" )
job.results.last_lattice_state().plot()
job.results.plot_molecule_numbers( ["CO2"], normalize_per_site=True )
job.results.plot_molecule_numbers( ["CO2"], normalize_per_site=True, derivative=True )
# xCO=0.55
job = pz.ZacrosJob.load_external( path="plams_workdir/plamsjob.035" )
job.results.last_lattice_state().plot()
job.results.plot_molecule_numbers( ["CO2"], normalize_per_site=True )
job.results.plot_molecule_numbers( ["CO2"], normalize_per_site=True, derivative=True )
Note
The code described in this tutorial can be significantly reduced using the built-in ZiffGulariBarshad model:
import scm.pyzacros.models
zgb = pz.models.ZiffGulariBarshad()
The model data can then be accessed by using zgb.lattice, zgb.mechanism, and zgb.cluster_expansion.
Additionally, the code taking care of individual executions of the ZacrosJob objects and recovering the results for each condition can also be simplified by using the Extended Component ZacrosParametersScanJob.
Take a look at the example PhaseTransitions-v2.py for further details.