H-NMR spectrum with spin-spin coupling¶
In this tutorial, you will learn how to:
use the GUI to set up the calculation of NMR chemical shifts and nuclear spin-spin coupling constants (NSSCCs)
use the AMSspectra module to inspect the results and compare the simulated and experimental NMR spectra directly
See also
Don’t miss this must-read FAQ entry on NMR settings based on the work done by the group of Jochen Autschbach
The NMR part of the ADF manual contains a neat summary of the science behind spin-spin coupling calculations as well as some additional advice.
The advanced tutorial on relativistic NMR calculations .
Start AMSinput and copy the molecule¶
O 1.48603879 -1.49561627 0.00000000
C 1.29751002 -0.30552432 0.00000000
O 0.07403584 0.25228979 0.00000000
C -1.02449892 -0.67494471 0.00000000
C -2.30056502 0.12358769 0.00000000
C 2.36905363 0.74347075 0.00000000
H -0.94187587 -1.31519741 0.88039373
H -0.94187587 -1.31519741 -0.88039373
H -2.36617127 0.75820872 0.88525259
H -3.15628689 -0.55419212 0.00000000
H -2.36617127 0.75820872 -0.88525259
H 3.34355252 0.26293272 0.00000000
H 2.26362714 1.38098693 0.87932777
H 2.26362714 1.38098693 -0.87932777
The above structure was optimized with the following settings:
Hybrid XC: PBE0
Basis: TZP
Frozen core: None
Numerical Quality: Good
In case you want to run the geometry optimization yourself, take a look at the GUI tutorial on geometry optimizations.
Setting up the NMR calculation¶
Select the following settings in AMSinput
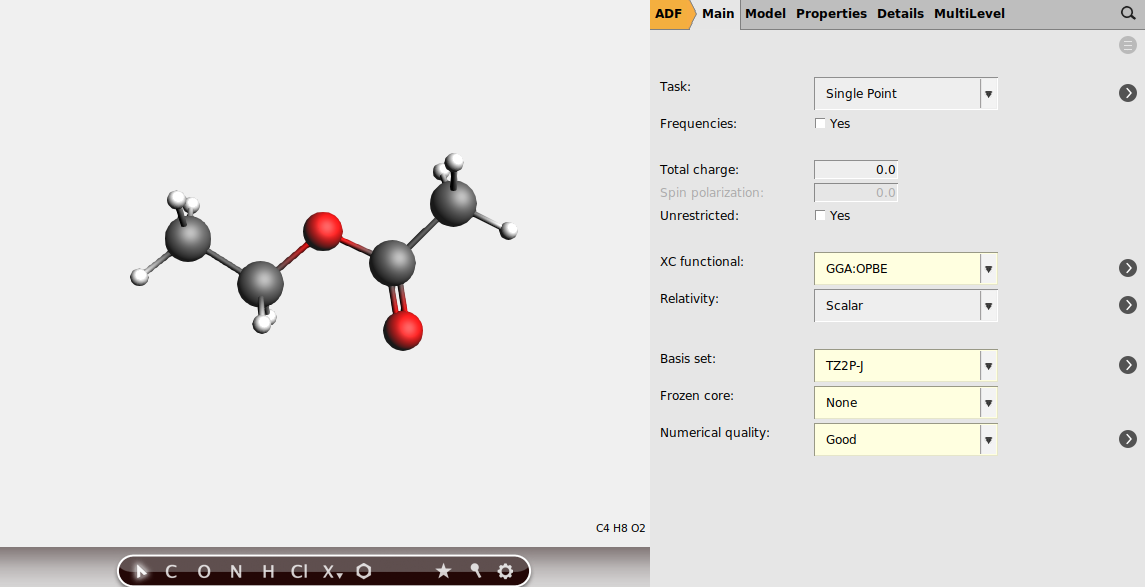
Note
The basis sets in J, including TZ2P-J, have been especially designed for ESR hyperfine and NMR spin-spin coupling calculations.
Next, instruct the program to calculate the shieldings for all hydrogen atoms.
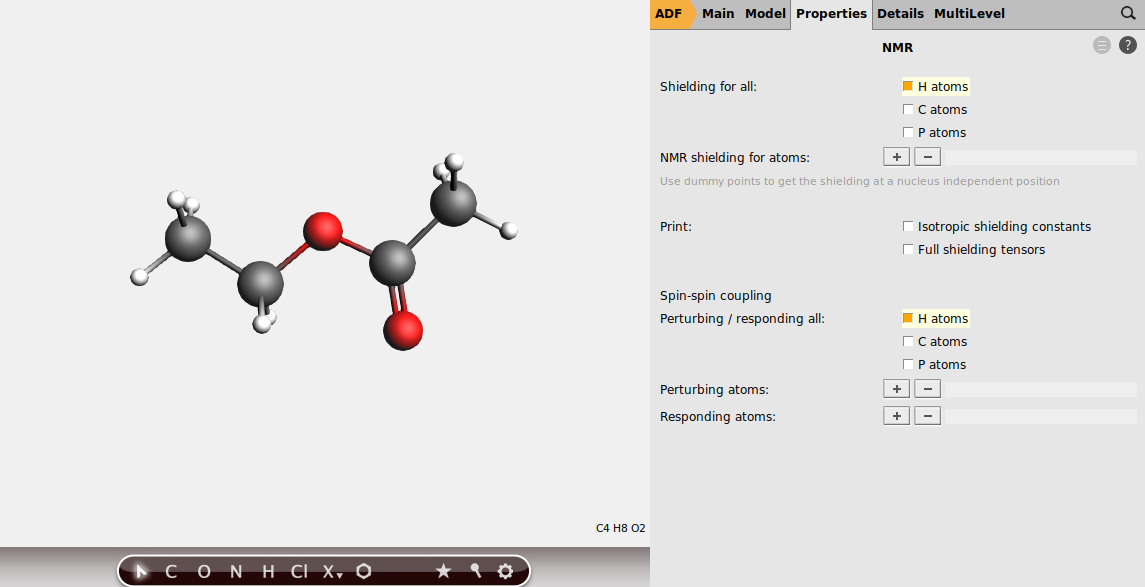
Note
In some cases, e.g. when dealing with alcohol groups, you might want to exclude atoms from the list of perturbing and responding atoms. To do so, just select the atoms you want to calculate the splittings for, and use the + button to add to the list of perturbing and responding atoms manually.
You have now finished the setup of the calculation and are ready to run it.
Results of your calculations¶
Logfile: AMStail¶
You can follow the progress of the calculation by clicking SCM → Logfile (AMStail). The chemical shifts will be calculated first, followed by the couplings constants for the perturbing and responding atoms.
When the calculation has finished the end of your logfile will look something like this:
View the 1H-NMR spectrum¶
Use AMSspectra to visualize the calculated spectrum
The NMR chemical shift \(\delta_i\) is defined as the difference of the isotropic NMR chemical shielding of a reference compound \(\sigma_{ref}\) minus that of the interesting compound \(\sigma_i\):
The NMR program calculates NMR chemical shieldings \(\sigma_i\). AMSspectra uses as 1H reference (TMS) shielding \(\sigma_{ref}\) = 31.7 ppm.
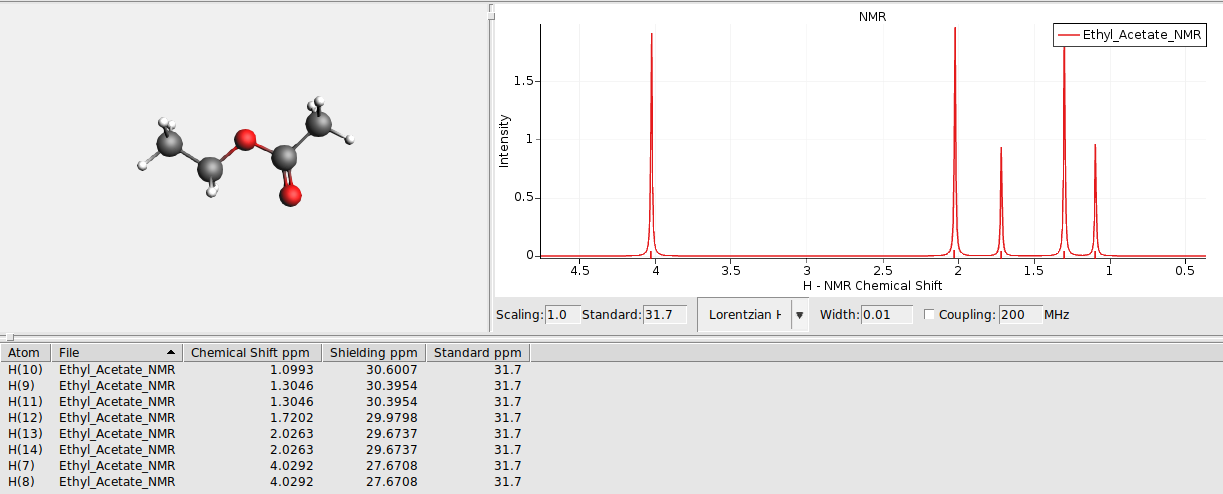
By default only the chemical shifts are visualized using plain singlets. To switch on the visualization of couplings:

This switches on the spin-spin coupling (default machine frequency at 200 Mhz). An additional section in the table will appear which displays additional information for any selected atom or peak in the spectrum. For example:
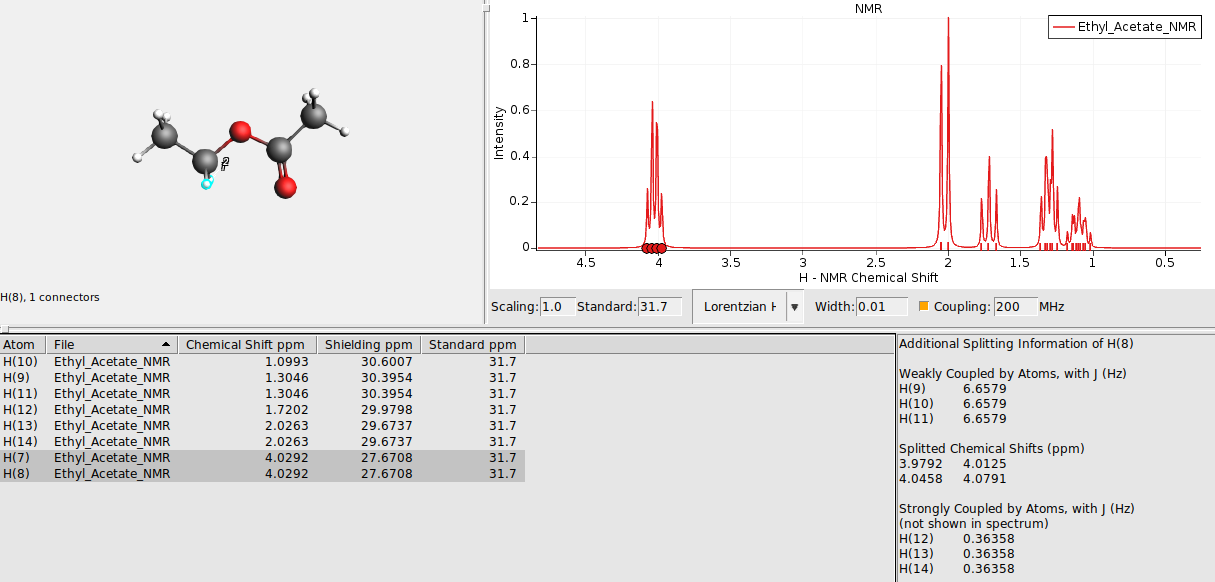
Average chemical shifts and couplings for equivalent atoms¶
As you may have noticed, there are a lot of splittings in the simulated spectrum. This is because all the chemical shifts and coupling constants are calculated at fixed geometry, which means that there is no rotation which would create magnetically equivalent groups.
You can resolve this issue by either flagging chemically equivalent atoms manually or have AMSspectra guess them for you. To manually supply the information:
Select groups of chemically equivalent atoms and
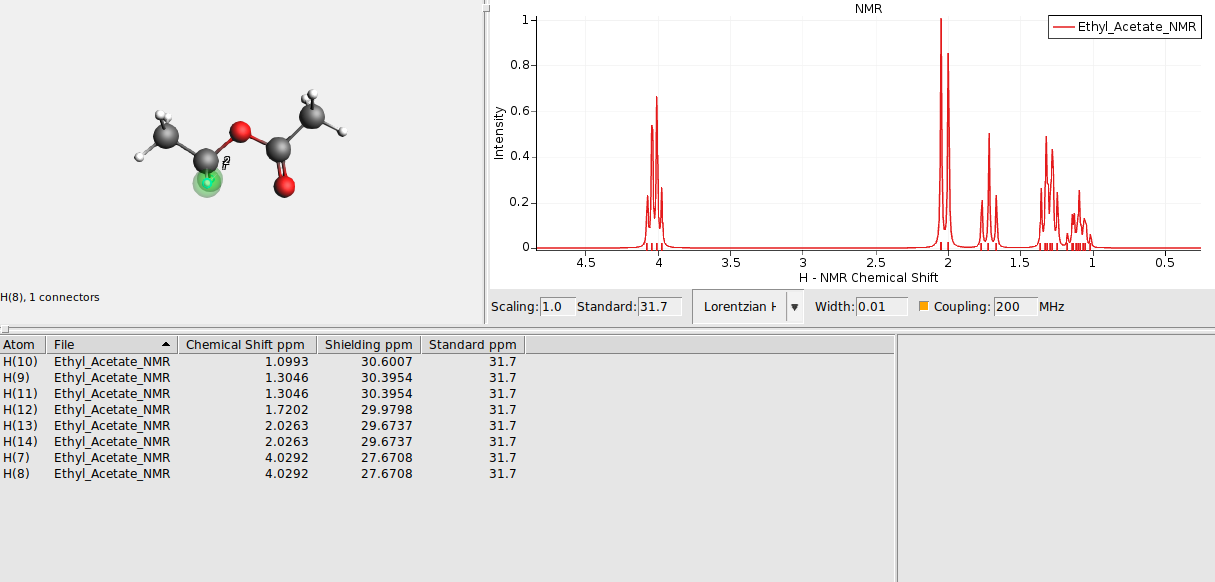
The atoms should now be surrounded by a colored sphere. Continue with all remaining atoms. Your spectrum should look as follows now:
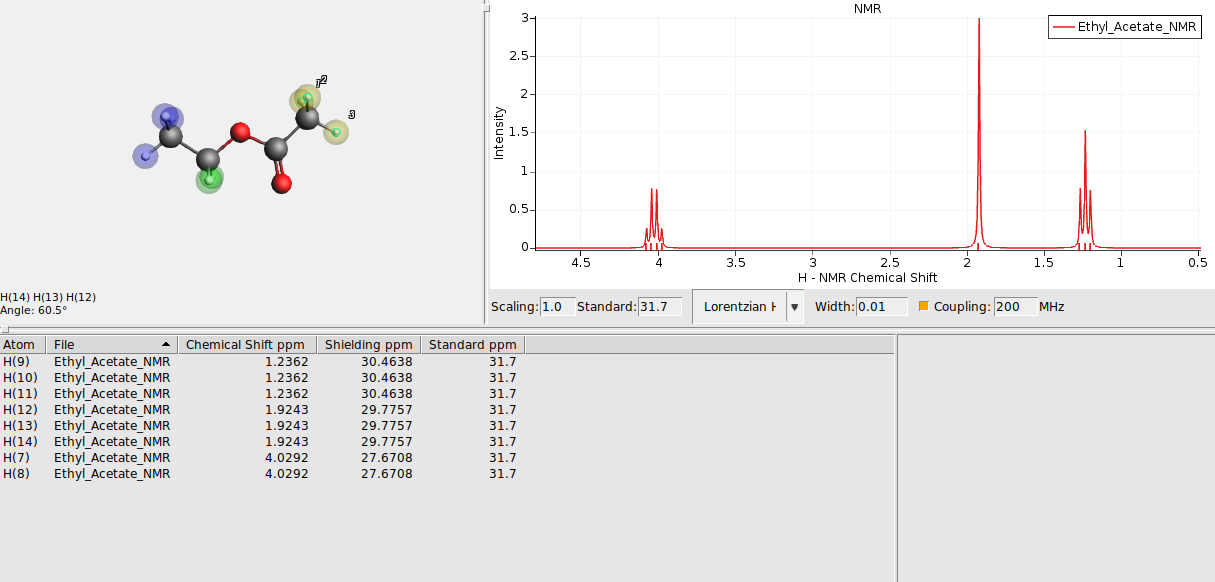
Alternatively, to have AMSspectra guess chemically equivalent regions for you, go to
Tip
In the NMR menu you can adjust the thresholds that are used by the algorithm to identify equivalent regions.
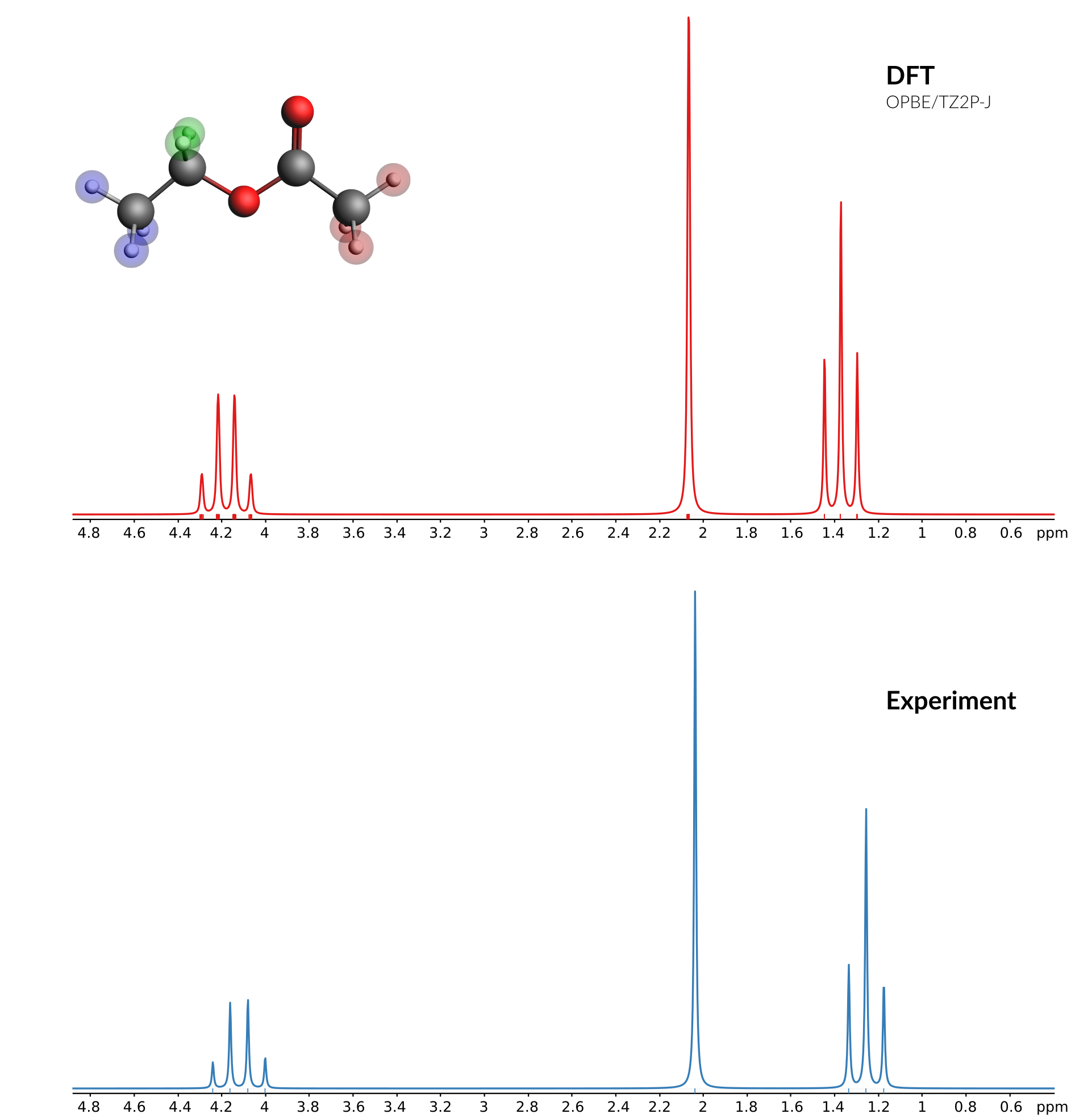
Comparison of calculated and experimental spectrum¶
A good source for experimental spectra is the SDBS database: http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
These are the peak data for the 1 H-NMR spectrum of ethyl acetate (CAS = 141-78-6) from the SDBS database (the three columns are Hz, ppm and Intensity):
379.88 4.242 52
372.75 4.163 171
365.56 4.082 180
358.50 4.003 62
182.56 2.039 1000
119.69 1.337 245
112.63 1.258 560
105.38 1.177 214
Now we switch back to AMSspectra where we can paste the contents from the clipboard directly:
AMSspectra will ask which columns to use for X and Y, and offer to rescale the data:
2 to use the second column as X values (that column contains the chemical shift in ppm) and click OK3 to use the third columns as Y vales and click OKYou now should see both the experimental and the calculated spectrum in one graph. The experimental spectrum (at least the one used to create this tutorial) was measured at 89.56 MHz. So adjust the frequency (default at 200 MHz) of the calculated spectrum:
89.56 MHzThis should give you something like this:
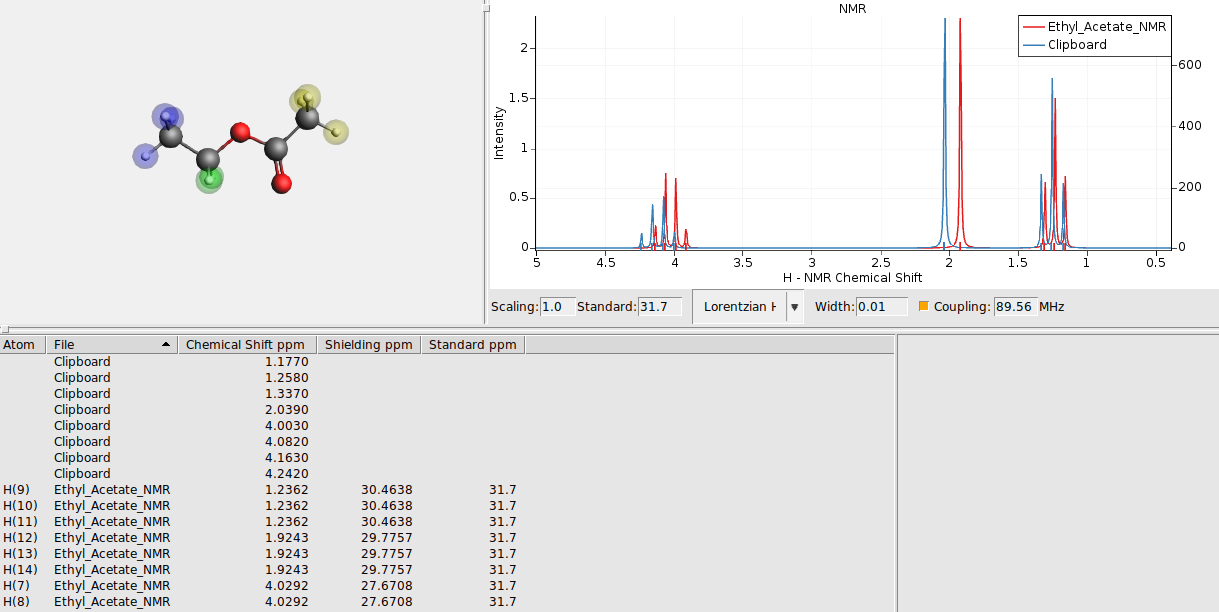
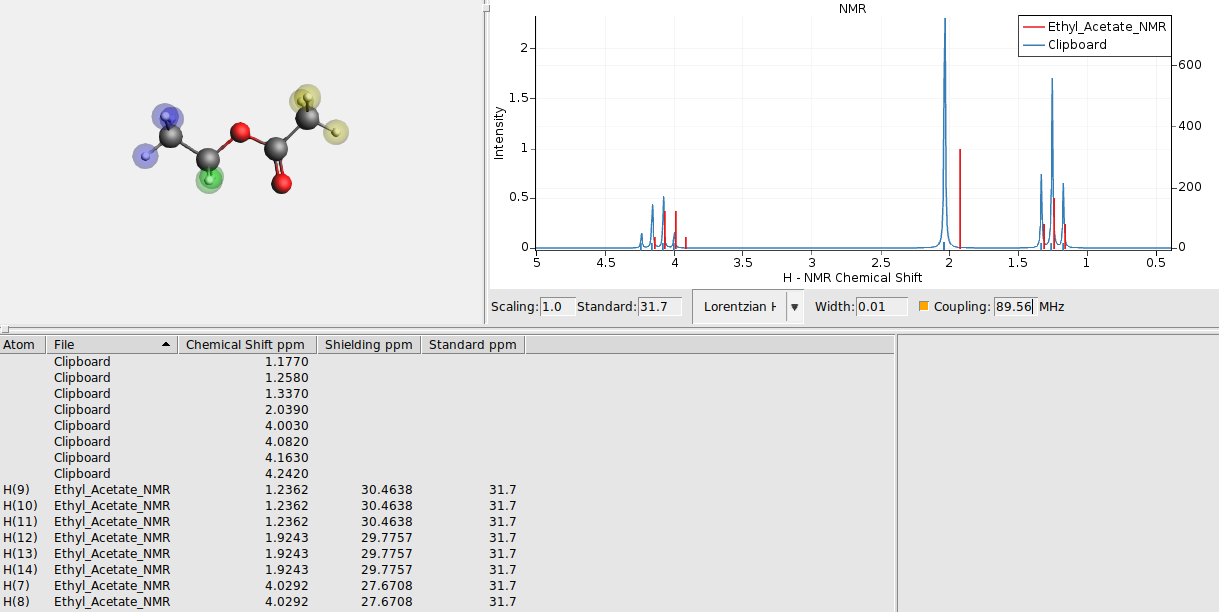
Sometimes it makes sense to change one of the spectra to a stick spectrum, showing only sticks for the calculated positions and heights, not applying the broadening. As an example lets do that here for the calculated peak positions:
Now you should get the experimental spectrum as a broadened curve, with sticks for the calculated positions:
Spectrum overlap¶
If you have an (experimental) spectrum in xy format with the same units as your calculated spectrum, you can calculate and optimize the overlap. The method used to calculate the overlap is SimIR/VCD from J. Shen et al. Spectrochimica Acta Part A 76 (2010) 418-422. First, we start with a clean AMSspectra window with our NMR calculation and apply the previous NMR options:
89.56 MHzNow we will add an experimental spectrum that has the same units as our calculated spectrum:
here to download the .xy file Ethyl_acetate.xyTip
If your spectrum has different units you will want to put it on the right axis, so you can scale it separately. In that case you cannot calculate or optimize the overlap.
We will now optimize the overlap. The peak width, standard reference (or offset in general), and scaling will be optimized to maximize the overlap between the spectra.
Important to note is that only the current horizontal range is used to optimize the overlap. Specific areas of the spectrum can be optimized this way by limiting the range. The results are immediately applied to the calculated spectrum. If the spectra are too dissimilar the automatic optimization will fail, therefore first the calculated spectrum is shifted by changing the reference chemical shift, such that the largest peak at around 2 ppm almost coincides.
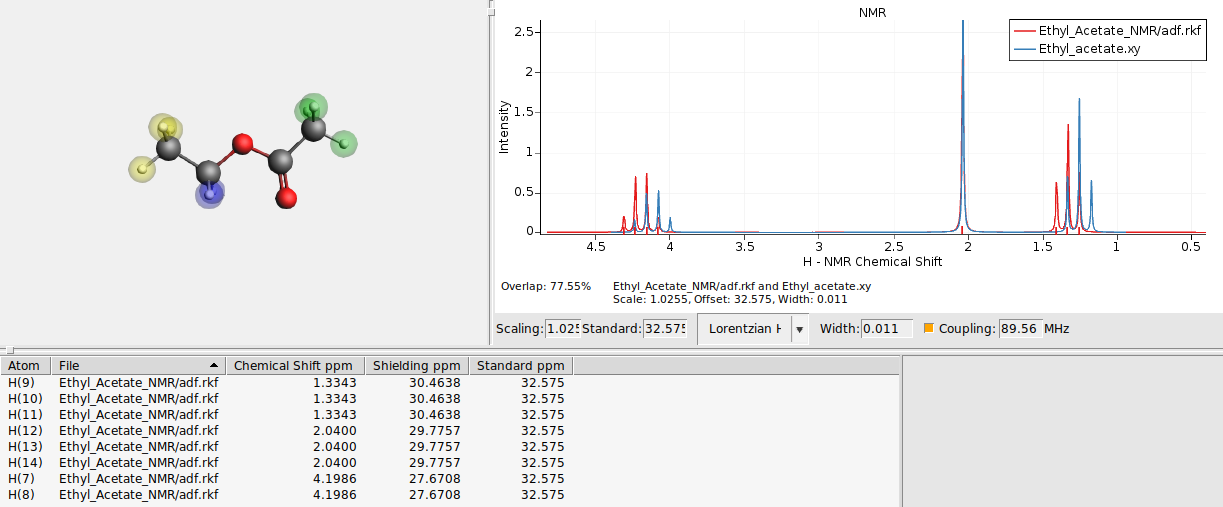
The automatic optimization is sensitive to numerical noise, thus the results of the automatic optimization may be different than the shown results. Instead of the automatic optimizing of the overlap, just the overlap can be shown and updated on the fly when the width, standard, scale or horizontal range are changed.