Quantum ESPRESSO: Phonon Band Structure, DOS, and Thermodynamics¶
The phonon band structure and density of states (DOS) are important concepts in solid-state physics, which provide crucial insights into the materials vibrational properties. These concepts are essential for understanding various phenomena, including thermal conductivity, heat capacity, and electron-phonon interactions. From one side, the phonon’s band structure maps the allowed vibrational frequencies as a function of the wave vector, revealing how phonons propagate and interact within the crystal lattice. On the other side, the DOS measures the number of vibrational modes at each frequency. So, both create a complete picture of the vibrational properties. Additionally, from the phonon DOS, we can derive key thermodynamic properties such as internal energy, heat capacity, Helmholtz free energy, and entropy. These properties are essential for understanding the thermal behavior of materials, allowing us to predict and explain their macroscopic behavior. This understanding could also guide the design of materials with customized thermal properties.
Beryllium oxide (BeO), also known as bromellite, is a wide band gap (7.46 eV) semiconductor. It has a wurtzite crystal structure (space group \(P6_3mc\), No. 186) and exhibits interesting optical properties, making it relevant for applications in catalysis, high-power electronics, and neutron moderators in nuclear reactors. We obtained the system’s geometry from The Materials Project (ID: mp-2542). From the computational point of view, this system is relatively simple (only 4 atoms per unit cell) and has a small number of electrons (20 electrons per unit cell), which leads to faster calculations, making it suitable for illustrative purposes. Theoretical results for comparison can be found in The Computational Raman Database (mpid: mp-2542).
Computational Methodology¶
The calculation of phonon properties using Quantum Espresso (QE) involves a series of steps that are handled internally by AMS, making the process convenient to the user. These steps use the QE tools pw.x, ph.x, q2r.x, and matdyn.x. For detailed information about these tools, refer to the official Quantum Espresso documentation online.
Starting from an optimized geometry, we perform a self-consistent field (SCF) calculation using pw.x to determine the electronic ground state of the system. This step solves the Kohn-Sham equations iteratively to obtain the converged electron density and ground state energy, which are essential for subsequent phonon calculations. Next, we use ph.x to calculate the phonon frequencies and eigenvectors at a uniform grid of q-points in the Brillouin zone. This is the most computationally expensive part. This step employs density functional perturbation theory (DFPT) to compute the dynamical matrix, which captures the interatomic force constants and provides the foundation for phonon analysis. Subsequently, q2r.x is used to perform a Fourier transform of the interatomic force constants from the q-point grid to real space, enabling the calculation of phonon properties in a more convenient representation. Finally, matdyn.x is employed to post-process the phonon data, allowing us to extract the phonon DOS and band structure (these are two independent calculations). This step involves calculating phonon frequencies for arbitrary q-vectors, including those along high-symmetry paths, using the interatomic force constants obtained from the dynamical matrices.
Once the phonon DOS \(D_p(\varepsilon)\) is calculated, we can determine various thermodynamic properties using the canonical distribution from statistical mechanics for phonons under the harmonic approximation. These properties are crucial for analyzing the thermal behavior of materials.
In AMS, thermodynamic properties are automatically calculated when the DOS is requested, offering a convenient way to access this information. The properties derived from the phonon DOS are calculated as follows:
Internal energy (\(U\)): Represents the total energy of the system due to phonons.
Specific heat (\(C_V\)): Measures the amount of heat required to raise the temperature of the material by a certain amount at constant volume.
Helmholtz free energy: (\(F\)): Represents the thermodynamic potential that measures the useful work obtainable from a closed system at a constant temperature and volume.
Entropy (\(S\)): Measures the degree of disorder or randomness in the system.
Note
Please note that the total DOS for phonons in AMS sums to \(3n\), where n represents the total number of atoms in the unit cell, indicating the total number of phonons.
These thermodynamic properties provide a comprehensive picture of the thermal behavior of materials and are crucial for understanding and predicting their response to temperature changes. This information is particularly important for various applications, such as designing efficient heat sinks, thermal barrier coatings, and for understanding the stability and phase transitions of materials at different temperatures.
The phonons properties can be calculated using Quantum Espresso through AMS via two main interfaces: the graphical user interface (GUI) and the Python library, PLAMS. The GUI presents a user-friendly, visual approach that is ideal for users performing routine calculations on simple systems. It simplifies the process of setting up input files and visualizing results. On the other hand, PLAMS offers a powerful and flexible Python environment suitable for complex workflows, parameter sweeps, and integration with other scientific libraries. This makes it particularly advantageous for advanced users, high-throughput calculations, or situations that require automation and scripting, providing greater control and customization of the computational process. In this tutorial, we will explore both options. They are completely independent, so you can jump directly to the section that interests you the most.
Using the GUI¶
We will use the following structure as basis for the subsequent calculations:
Be 0.0000000000 1.5555413800 0.0007009000
Be 1.3471383600 0.7777706900 2.1887103500
O 0.0000000000 1.5555413800 1.6512462200
O 1.3471383600 0.7777706900 3.8392556700
VEC1 2.6942767100 0.0000000000 0.0000000000
VEC2 -1.3471383600 2.3333120800 0.0000000000
VEC3 0.0000000000 0.0000000000 4.3760188900
Hint
To build a custom crystal, check out the Tutorial on Building Crystals and Slabs
Setting Up the Calculation:

You should get something like this:
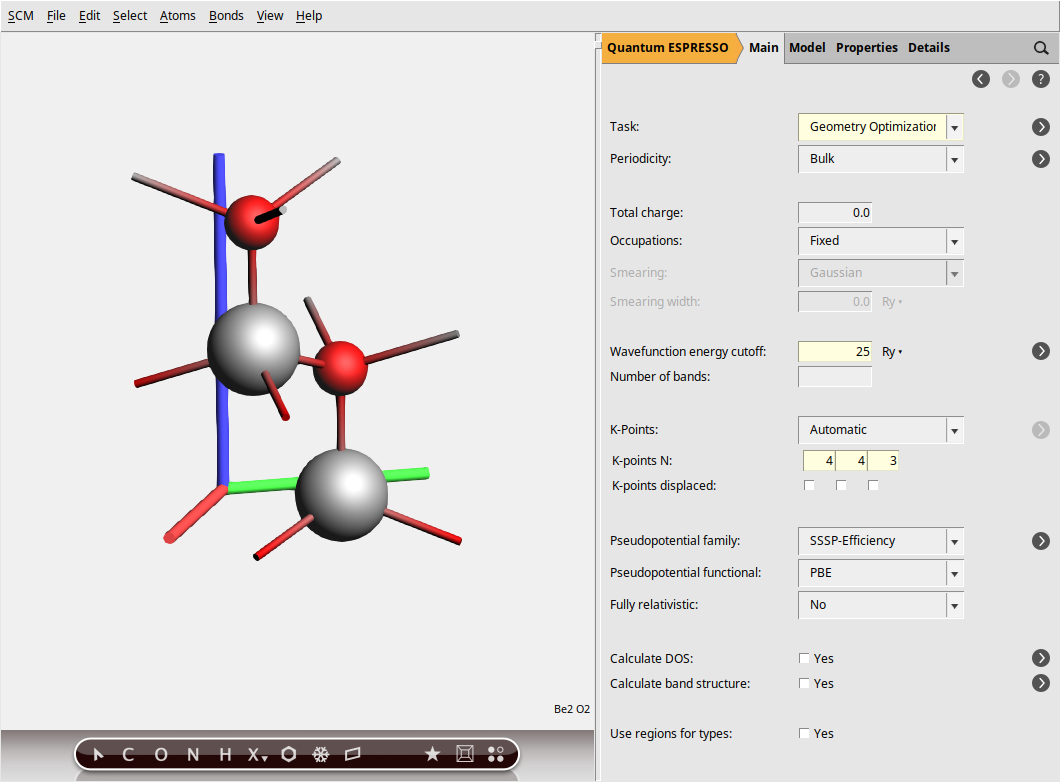
In the steps above, we specified a GeometryOptimization task to ensure our BeO structure is at a local energy minimum, preventing any spurious imaginary frequencies in the phonon spectrum.
Note
When computing phonons, you should in general optimize the lattice vectors as well. In this example, the lattice parameters are already adequate, so we skip the lattice optimization step. For more information, see the Lattice Optimization and Phonons tutorial.
Next, for the electronic structure part, we set the plane-wave energy cutoff (ecutwfc) to 25 Ry. While this value is lower than typically recommended for production calculations (40 Ry), we use it here for demonstration purposes to reduce computational cost. Then, the Brillouin zone sampling is done using a Monkhorst-Pack k-point grid of size 4 × 4 × 3, ensuring a balanced sampling in all directions.
Note
This calculation uses the default pseudopotentials, corresponding to the SSSP-Efficiency family with the PBE functional and no relativistic corrections.
Now, use the panel bar Properties → Phonons and Elastic Tensor command to go to the Phonons (and Elastic Tensor) configuration panel. Then:
 next to the Method entry to configure the details of the Analytical Phonons calculation.
next to the Method entry to configure the details of the Analytical Phonons calculation.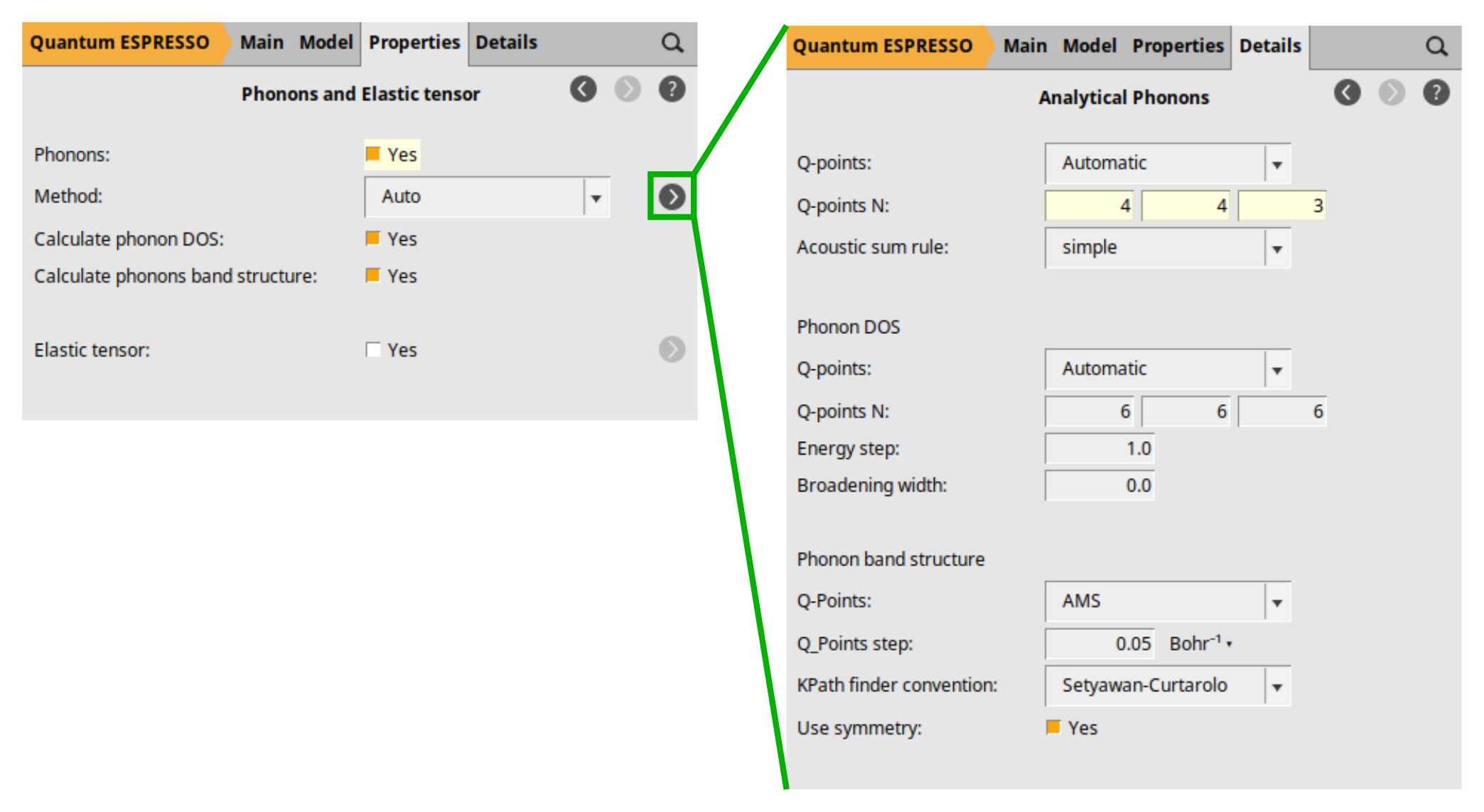
Here, we activated the calculation of phonons (Phonons: Yes), which by default calculates both the phonon band structure and the DOS. Notice the checkboxes Calculate phonon DOS and Calculate phonons band structure. We defined a 4 × 4 × 3 q-point grid for our phonons calculation. A coarser q-point grid is normally preferred to the k-point grid (electronic part), as phonon calculations are more computationally intensive. However, the right choice should be obtained by a convergence test. AMS uses the specified q-grid for the initial analytical calculation of phonon frequencies within Quantum Espresso. However, the band structure plot is generated by interpolating those frequencies onto a denser set of q-points along the high-symmetry path proposed by Setyawan and Curtarolo. You can change the path convention using the dropdown list KPath finder convention:. This interpolation scheme, using a Fourier interpolation, allows for the construction of continuous phonon dispersion curves, providing a clearer visualization of the phonon band structure and facilitating the analysis of vibrational frequencies across different regions of the reciprocal space.
Note
In the dropdown list labeled Method:, you can choose from Automatic, Numerical, or Analytical to select either numerical or analytical phonon calculations. For Quantum Espresso (QE), selecting Automatic (the default) is equivalent to set the Analytical.
Computation & Results¶
We are now ready to start the calculation
BeO)When you run a calculation, AMSjobs should come to the forefront, and your job will appear at the top of the list. On the right side, you’ll see the job running, indicated by a gear icon. While the simulation is in progress, the AMSjobs window will display the progress based on the logfile. Once the job is complete, the job status icon will change to a full circle to indicate that it is ready.
The AMSband module allows you to analyze both the Band structure and the DOS. Both properties are widely used because they reveals key features such as the distinction between acoustic and optical modes, the dispersion and anisotropy of phonon frequencies, the presence of frequency gaps, and the distribution of vibrational modes.
You should get the following:
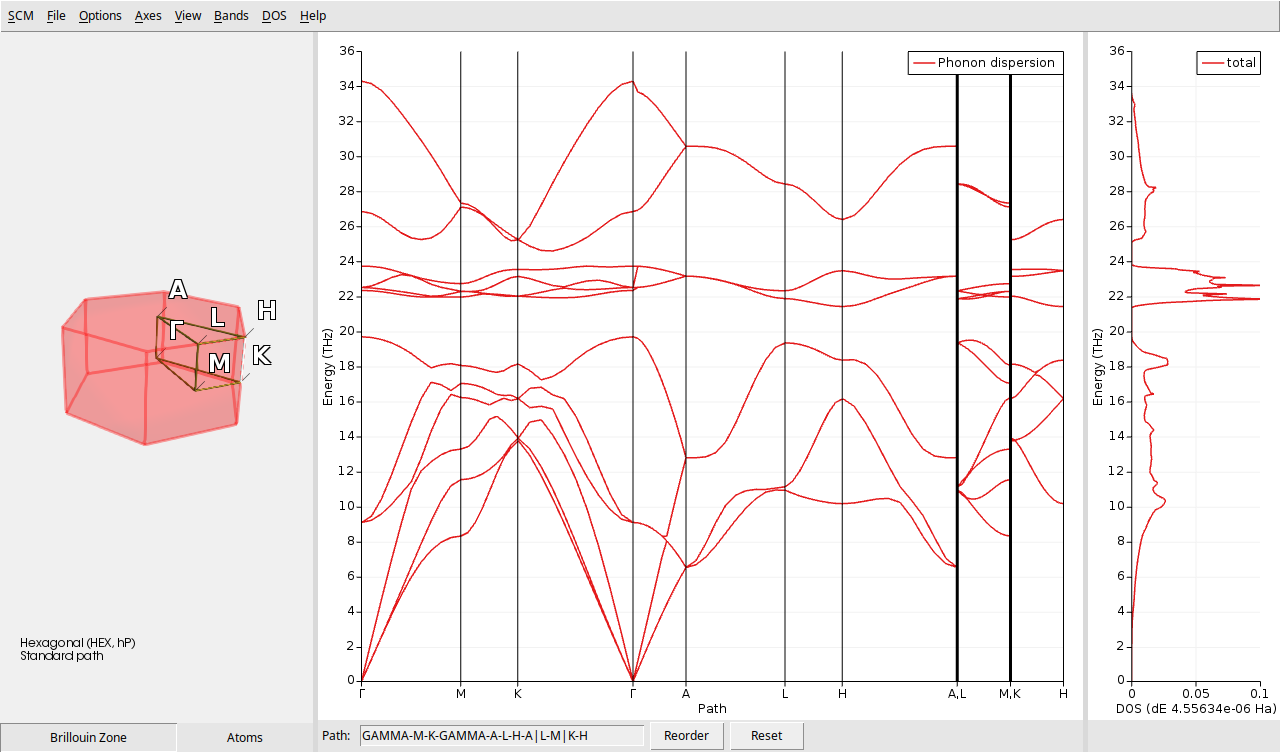
AMSbands provides a summarized view of the vibrational properties of BeO. On the left side, there is a visual representation of the first Brillouin zone. In the central section, you can see the phonon band structure, while the right side displays the DOS.
The Brillouin zone of BeO, shown on the left, reflects its hexagonal wurtzite crystal structure. Focusing on the central portion, the red lines indicate the dispersion of phonon frequencies across different high-symmetry directions in the Brillouin zone. Distinct branches correspond to acoustic and optical phonon modes: acoustic modes approach zero frequency at the center (\(\Gamma\) point), while optical modes exhibit non-zero frequencies. The right panel illustrates the phonon DOS, which indicates the number of vibrational modes at each frequency. Peaks in the DOS correspond to regions with a high density of phonon modes.
These combined plots offers insights into how phonon frequencies vary with the wave vector, highlights the dominant vibrational frequencies, and reveals the presence of any gaps or degeneracies in the phonon spectrum.
Theoretical results for comparison can be found in The Computational Raman Database (mpid: mp-2542).
Tip
In AMSbandstructure, you can freely move, rotate, and zoom the Brillouin Zone visualization to find the best viewing angle. For finer control, zoom in or out on individual axes on the bands or DOS diagrams by pointing your mouse cursor at the desired axis and using the scroll wheel. Double-click an axis to access its configuration window for even more precise adjustments, like changing ranges or the curves’ colors.
Tip
In AMSbandstructure, you can change the relative size of the three panels by clicking and dragging the gray line between the panels.
The AMSdos module allows you to focus on the DOS.
You should get the following:
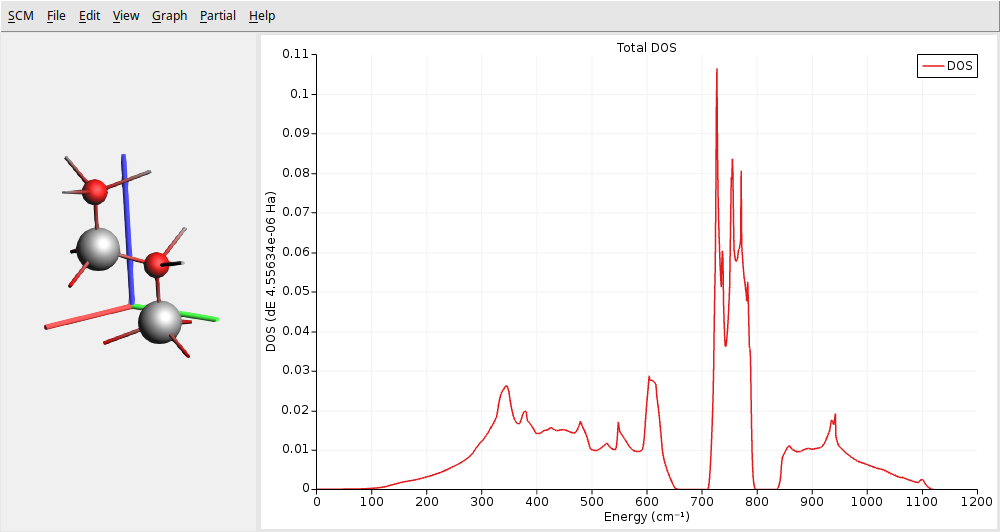
This figure shows three distinct regions separated by two gaps. At lower frequencies, the first region typically has contributions from acoustic and optical modes (confirmed with the phonon band structure above). Optical modes primarily dominate the second and third regions. In particular, we can see the intense characteristic peak associated with wurtzite structures around \(650 \text{cm}^{-1}\). This peak is mainly attributed to the \(E^2_h\) optical phonon mode, which is quite flat in the band structure. This mode involves vibrations perpendicular to the c-axis and is exclusively Raman-active.
After examining the phonon band structure and the DOS, we will now focus on the thermodynamic properties derived from the latter, as described at the beginning of this tutorial. To do this, we will use the KFBrowser tool:
KFBrowser allows you to explore all the data stored in the binary files ams.rkf (specific to AMS) and engine.rkf (specific to the engine). It contains all the information considered important during the entire calculation, such as energies, geometries, gradients, and in this case, the thermodynamic properties of the phonons. Following the instructions above, you will open the engine file quantumespresso.rkf, which should appear as follows:
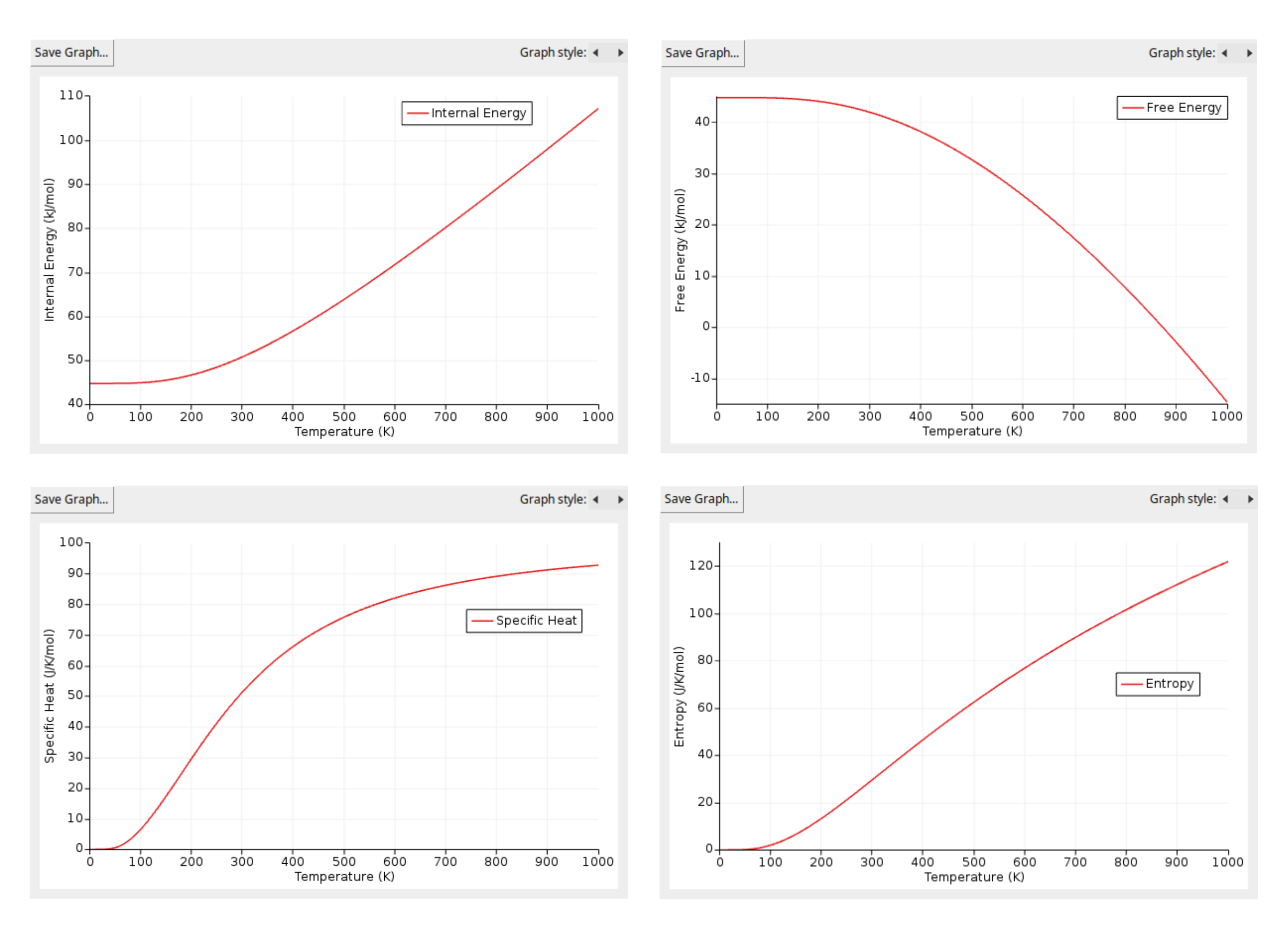
In the Plots section, you will find four thermodynamic properties: internal energy, free energy, specific heat, and entropy. You can generate a clear figure for each property by simply double-clicking on the one you are interested in. The figures you should obtain are the following:
Tip
In these plots, double-click on the y-axis to change the units, for example, from Hartree to kJ/mol for internal energy and free energy, or from kB (Boltzmann constant units) to J/K/mol.
These figures illustrate the calculated thermodynamic properties of BeO as a function of temperature, derived from the phonon DOS. From them, we can see a couple of interesting features:
Firstly, the internal energy (\(U\)) increases steadily with temperature, starting from the Zero Point Energy (ZPE) at 0 K (44.70 kJ/mol). This trend aligns with the increase in vibrational energy of the atoms as the temperature rises.
Secondly, the specific heat (\(C_v\)) exhibits a characteristic behavior commonly observed in many solids. It increases rapidly at low temperatures due to the gradual excitation of vibrational modes. As the temperature continues to rise, the specific heat approaches a plateau known as the Dulong-Petit limit. This limit represents the classical regime in which all vibrational modes are fully excited. For BeO, our calculations indicate that the Dulong-Petit limit is approximately 99.77 J/K/mol of unit cells, which corresponds to 3n*R, where n is the number of atoms per unit cell and R is the ideal gas constant. To express this limit per mole of atoms, as is traditionally done, we divide by n to arrive at the well-known value of 3R.
Using Python (PLAMS)¶
The full PLAMS workflow is now maintained as a scripting example:
It contains both downloadable notebook/script files and the rendered step-by-step Python workflow.
See also:
Quantum ESPRESSO IR and Raman spectra for BeO for the related IR and Raman workflow.