Quantum ESPRESSO: Geometry and Lattice Optimization¶
This tutorial will guide you through the following steps:
Setting up a geometry optimization (including lattice) of a solid using the Quantum ESPRESSO engine in AMS.
Viewing the results using AMSmovie.
Optional: Calculating and visualizing the band structure and partial density of states (DOS).
Note
Starting with AMS2024, there is a new engine in AMS called Quantum ESPRESSO. This engine launches the open-source Quantum ESPRESSO programs.
This means that the input is prepared in the normal AMS format. It is then internally converted to the Quantum ESPRESSO format.
Step 1: Start AMSinput and make sure Quantum ESPRESSO is installed¶
If this is your first Quantum ESPRESSO calculation using AMS, you must first install Quantum ESPRESSO. You can easily download and install it using the AMS package manager.
 →
→ 
Step 2: Set up the system - Silicon¶
ctrl+v) into the molecule-drawing area of AMSinputSi -0.67875000 -0.67875000 -0.67875000
Si 0.67875000 0.67875000 0.67875000
VEC1 0.00000000 2.71500000 2.71500000
VEC2 2.71500000 0.00000000 2.71500000
VEC3 2.71500000 2.71500000 0.00000000
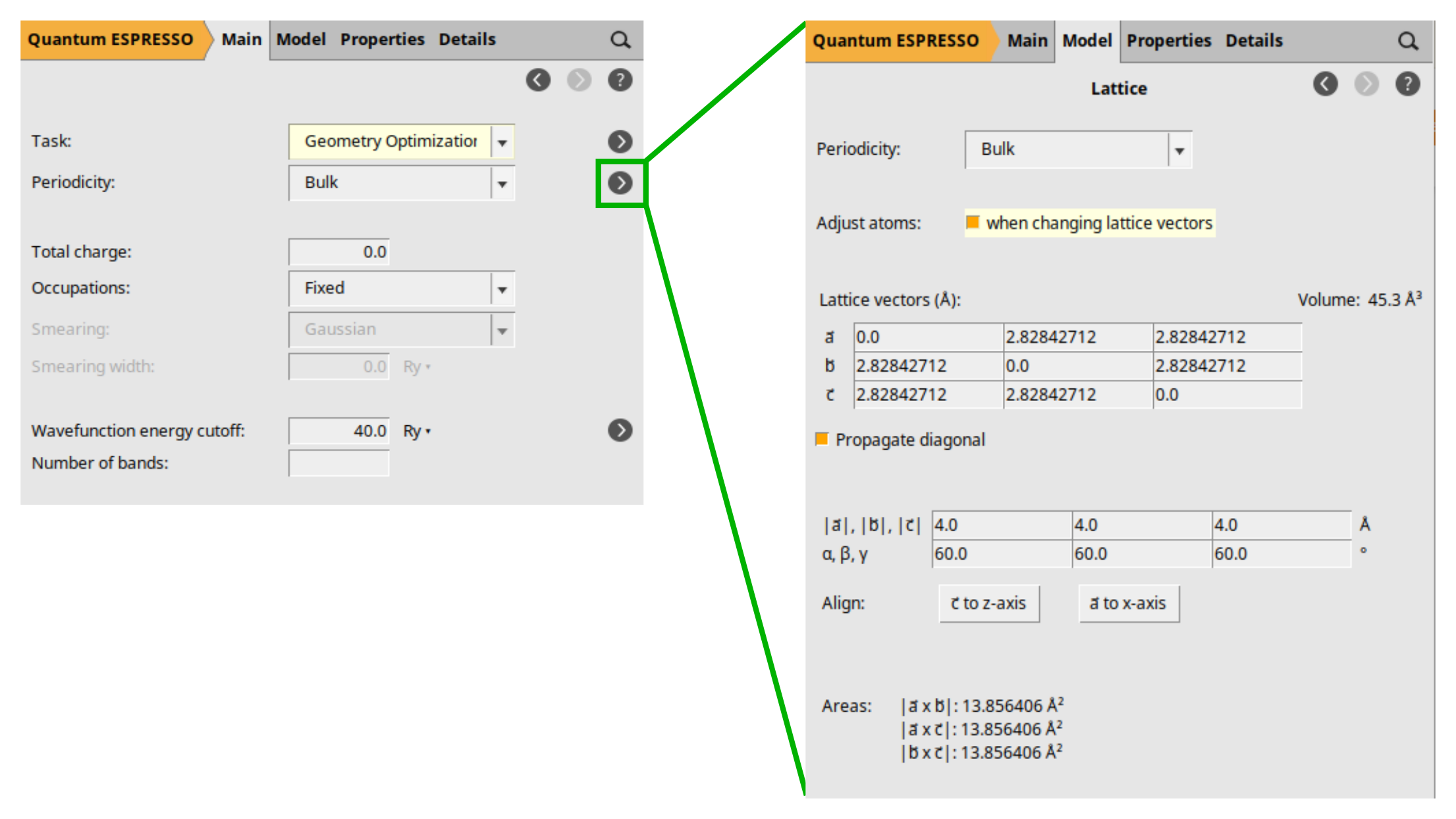
As we want to demonstrate lattice optimization, we will make slight changes to the lattice to move the system away from equilibrium. However, to ensure that the system’s symmetry is maintained throughout the optimization process, we will change the lattice vector lengths instead of their components directly, and we will allow the automatic adjustment of the atomic positions in response to these changes in the lattice vectors:
 button to the right of the Periodicity option
button to the right of the Periodicity option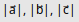
Note
The order of steps 2 and 3 is crucial. Switching them, i.e., adjusting the vectors’ lengths before allowing automatic atom adjustments, will result in undesired atom coordinates updates, leading to broken symmetry. This broken symmetry will induce wrong results on certain properties, e.g., the band structure.
Step 3: Setting up the calculation¶
First set up the required geometry optimization:
button to go to the optimization details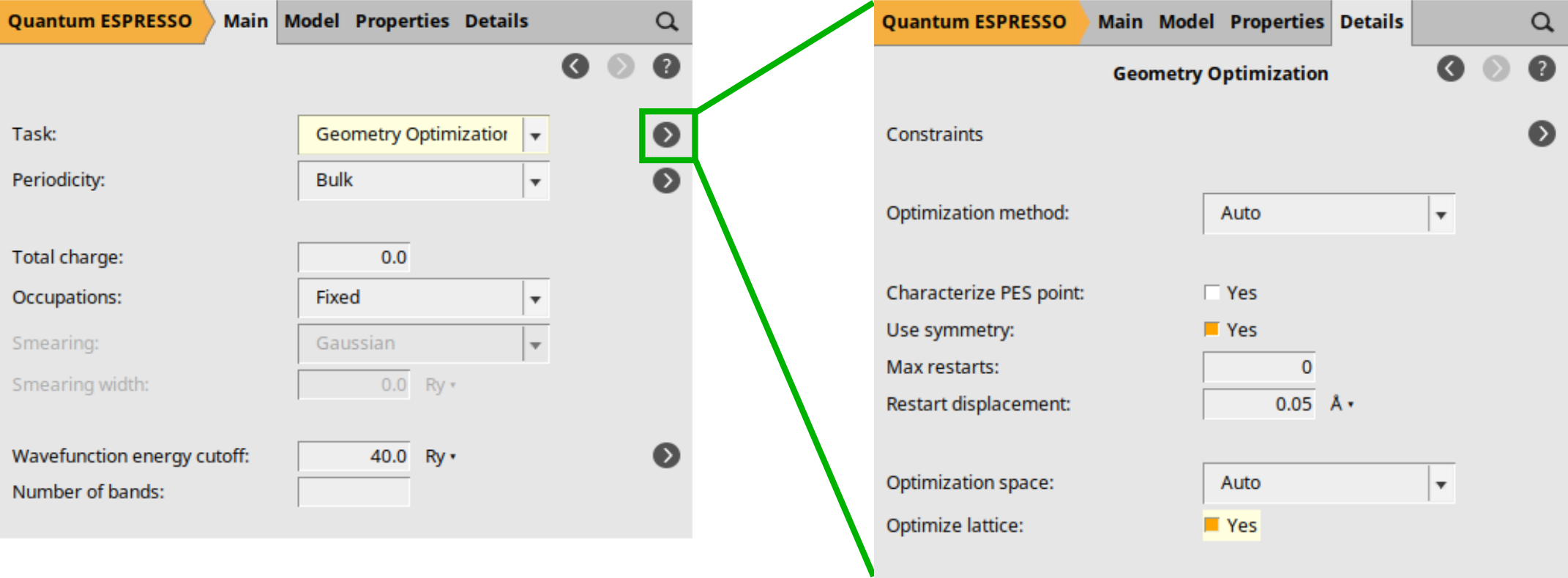
We also need to set up the general calculation options.
For this tutorial we will leave most options at their default values.
For real-life application you will have to check if your results are converged with respect to the k-points grid and wavefunction energy cutoff.
Starting with AMS2024, you have the option to use Pseudopotential Families, which significantly simplifies the process of selecting the pseudopotential that best fits your requirements. The three options that follow the Pseudopotentials title work as a filter: all pseudopotentials from the pseudopotential directory that are available for the elements in your system are shown. By choosing options (family, functional, and fully relativistic) you narrow down the list of available pseudopotentials.
If you click on the button located to the right of the Pseudopotentials family, you will be directed to the Pseudopotentials selection details.
In this section, you can view the specific UPF file that will be used in the calculation after filtering, along with the recommended minimum energy cutoffs for wave function and density.
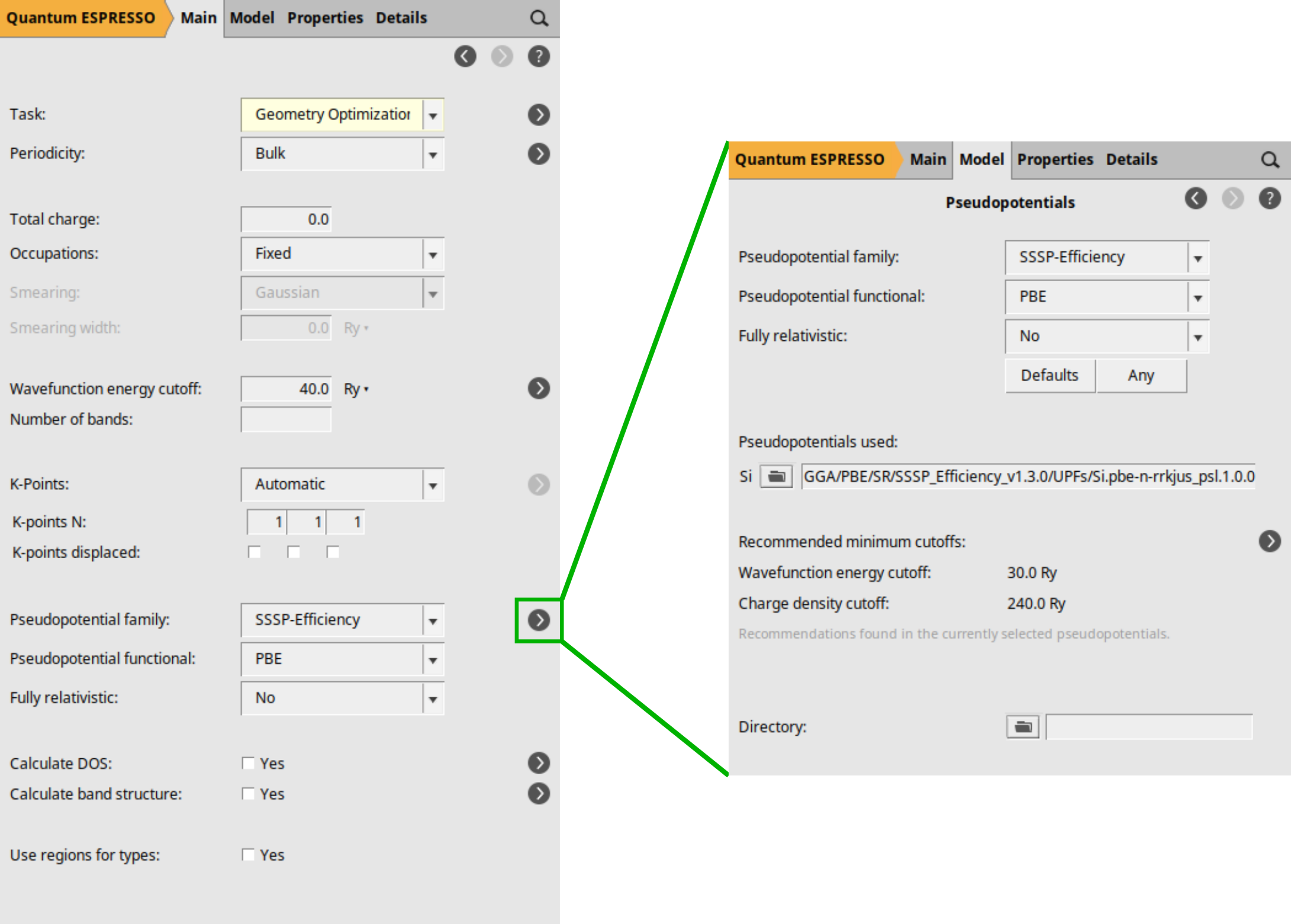
This is just an example, you will need to determine yourself (through calculation and/or literature) which pseudopotentials are best suited for your work.
Finally, set an appropriate k-space sampling (see k-space sampling for some recommendation). In Quantum ESPRESSO, an “automatic” k-point grid means a regular grid (Monkhorst-Pack grid).
10, 10, 10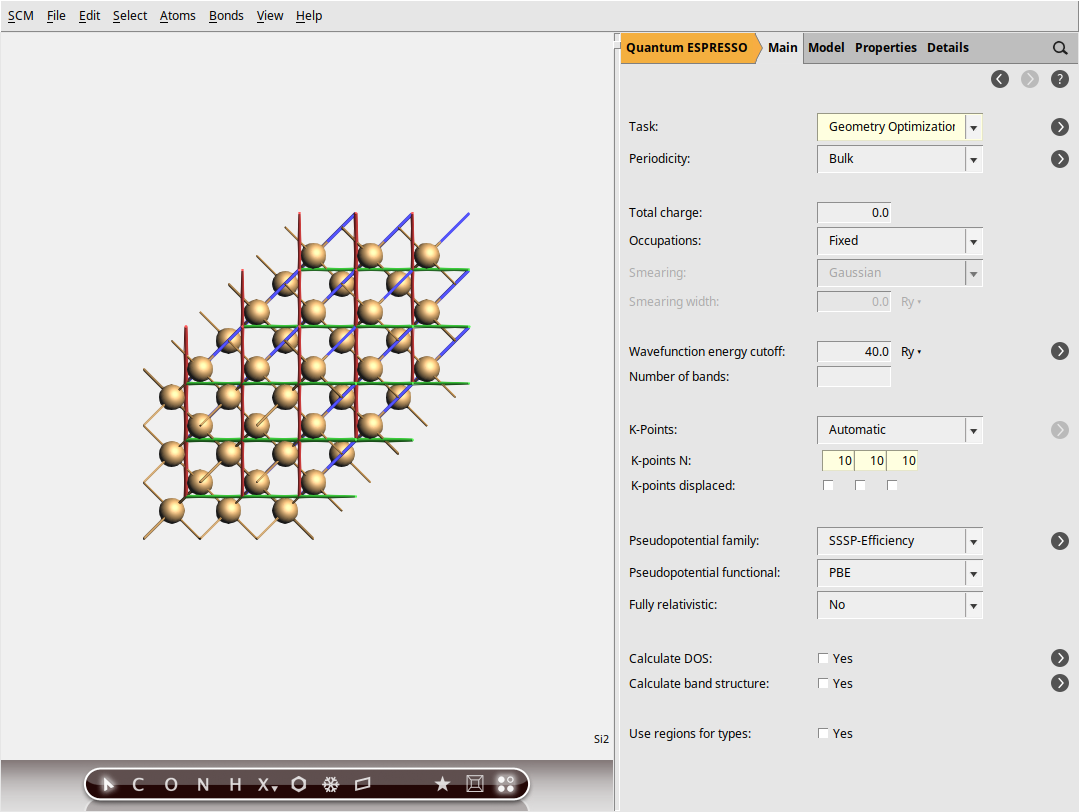
Step 4: Running your job¶
Running your job on your local machine is very simple:
silicon-geometryYour job should start running. In the AMSjobs window you can follow its progress, click on the progress lines to open AMStail showing full details of the progress.
When your job is ready AMSjobs will show this, and an Update molecule button will appear at the top of the Main panel in AMSinput. If you click this button, the geometry (both the atom coordinates and the lattice vectors) will be updated with the results of your calculation. Now you could continue to perform any other calculation on the optimized system using Quantum ESPRESSO or other AMS engines (like BAND).
Step 5: Checking the results¶
At the end of the calculation a .rkf file is automatically created based on the results of Quantum ESPRESSO. RKF files are used by the programs of the AMS suite,
We can now use all the GUI modules to examine the results.
In this tutorial we will have a look at the geometry optimization using AMSmovie:
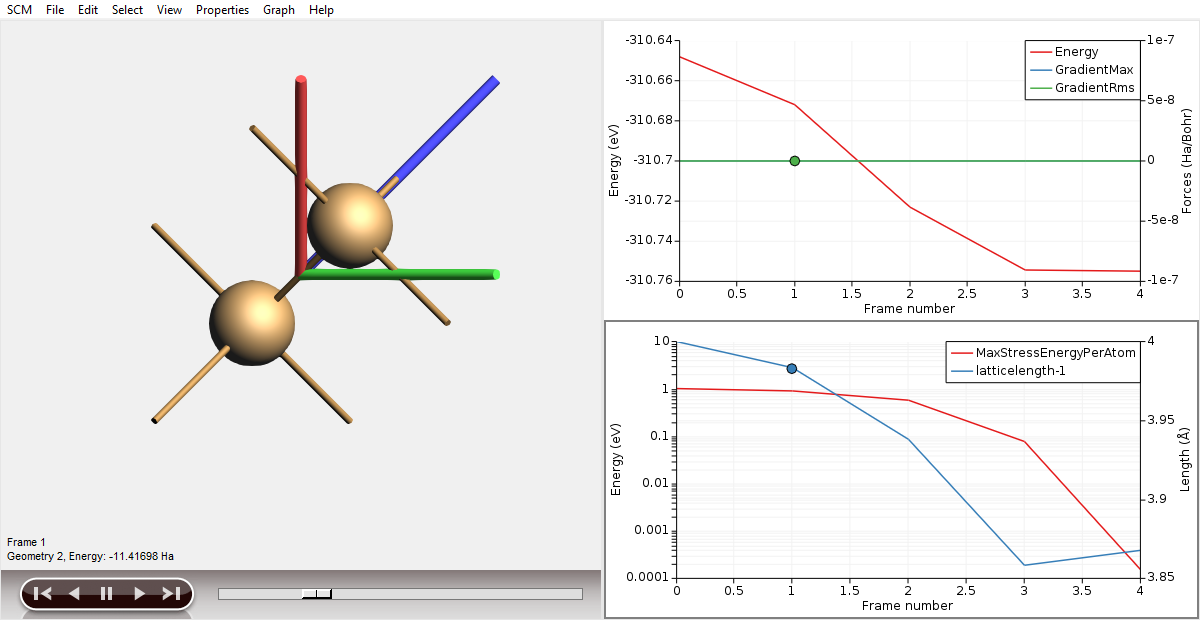
Step 6 (optional): Calculating band structure and partial DOS¶
silicon-properties: File → Save As… button located to the right of the Calculate DOS. you will be directed to the DOS selection details. There, check the Calculate PDOS and Lowdin charges check box.quantumespresso.rkf result file located in the folder named silicon-geometry.results.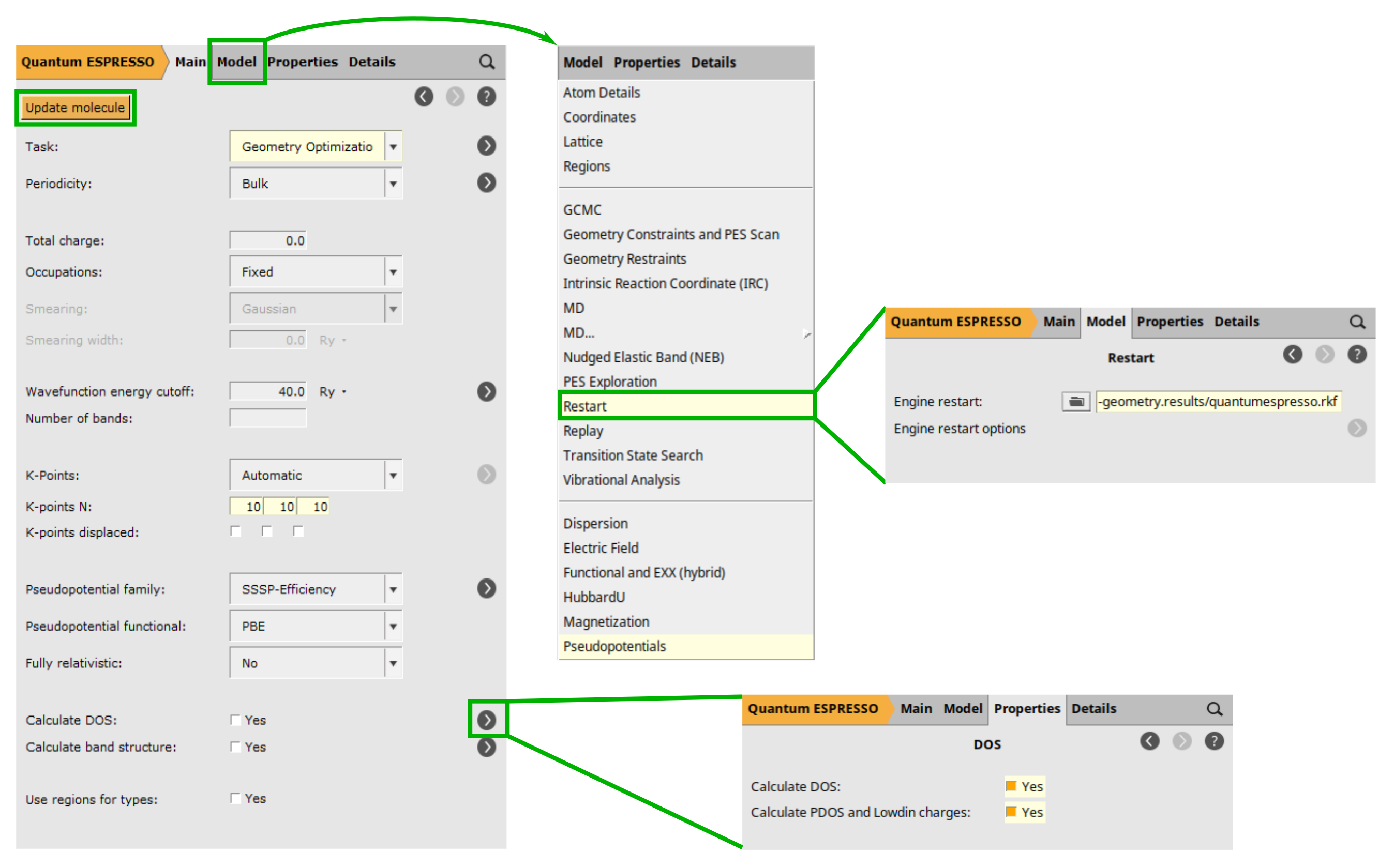
Your job should start running. In the AMSjobs window you can also follow its progress as in the previous calculation. In the AMSjobs window, note that the job status icon has changed (full circle) to indicate that the job is ready.
Step 7 (optional): Visualizing band structure and partial DOS¶
At the end of the calculation, an RKF file is automatically generated based on the Quantum ESPRESSO properties. Now, we can utilize the GUI modules to explore the results. Specifically, we will use AMSbandstructure to visualize the band structure and partial density of states (DOS) by following these steps:
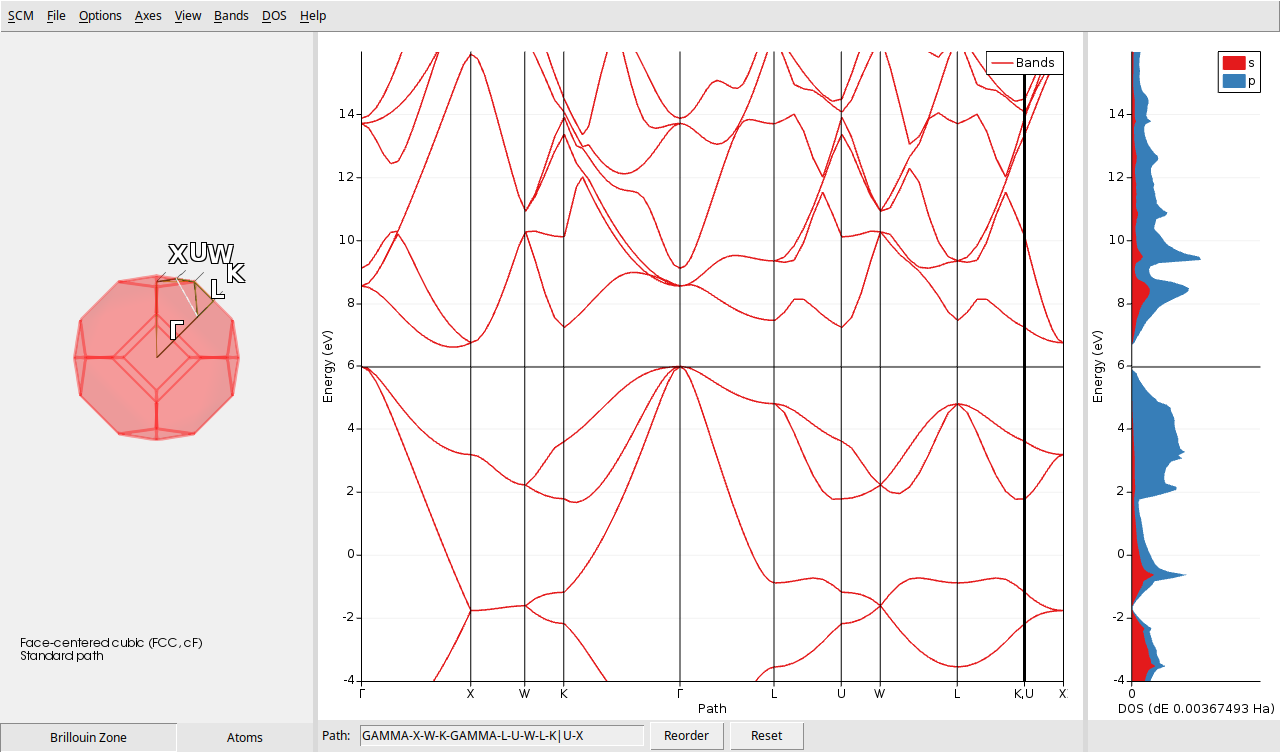
The AMSbandstructure window exhibits three linked plots. The left plot illustrates the Brillouin zone (BZ) space, representing the boundaries of the crystal’s unique vectors. The center plot depicts the band structure, where lines delineate the permissible electron energies relative to their wavevector, and the right plot displays the partial density of states (PDOS), featuring distinct curves representing the contributions of various electronic atomic shells to the available energy states.
Note
There is no need to divide the calculation into two steps (geometry optimization and property calculations). We did it here to demonstrate that this separation is possible. Because, in some cases, either of these steps can be very computationally demanding. So you can focus your efforts on geometry optimization and, later on, calculating different properties. However, keep in mind that you can enable property calculations directly from the first step. In this case, AMS will calculate the properties at the end using the optimized geometry.