VASP: TiO₂ surface relaxation¶
This tutorial will teach you how to:
Construct a slab for rutile TiO2(001)
Set up a geometry optimization job with constraints
Set up a DFT+U VASP job to be run via the AMS driver
See also
Tip
Download the GUI input filefor this tutorial (optional)
Step 1: Check the VASP installation¶
Important
VASP is not distributed with the Amsterdam Modeling Suite. The VASP program must have been obtained and installed separately.
Verify that you have access to a working installation of VASP 5 or VASP 6, on either your local or a remote machine. If you want to run on a remote machine or computer cluster, check that you have set up a working AMSjobs queue for that system.
Check that you can run VASP. An example command could be:
mpirun -np 4 vasp # parallelize over 4 cores
Depending on your system, the VASP command may be called vasp_gam or vasp_std.
If you do not know the proper command to launch VASP, ask your system administrator.
Step 2: Locate the POTCAR library¶
VASP requires the use of pseudopotentials or PAW potentials for each element. These are distributed with VASP in files called POTCAR or POTCAR.Z.
In this tutorial, you will use the Projector Augmented Wave (PAW) potentials for Ti and O constructed for the PBE density functional. You should have obtained those PAW potentials with VASP.
On your local machine (the machine running AMSinput), locate the needed POTCAR files. If you do not have them, you can download them from the VASP website.
For example, if the needed POTCAR or POTCAR.Z files are at
/some/path/PAW_PBE/Ti/POTCAR
/some/path/PAW_PBE/O/POTCAR
then the path /some/path/PAW_PBE/ would be the POTCAR Library that you need
to specify in AMSinput.
See the POTCAR documentation for the VASP engine for more details.
Tip
Consider saving your VASP command and POTCAR library as a preset (Right click any input field → Save Preset VASP). The next time you open AMSinput and select the VASP engine, this command and POTCAR library will be used.
Step 3: Create the TiO2(001) slab¶
 →
→ 
Note
One would normally perform a lattice optimization of the bulk structure before creating a slab. In this tutorial, we will instead use the experimental lattice parameters.
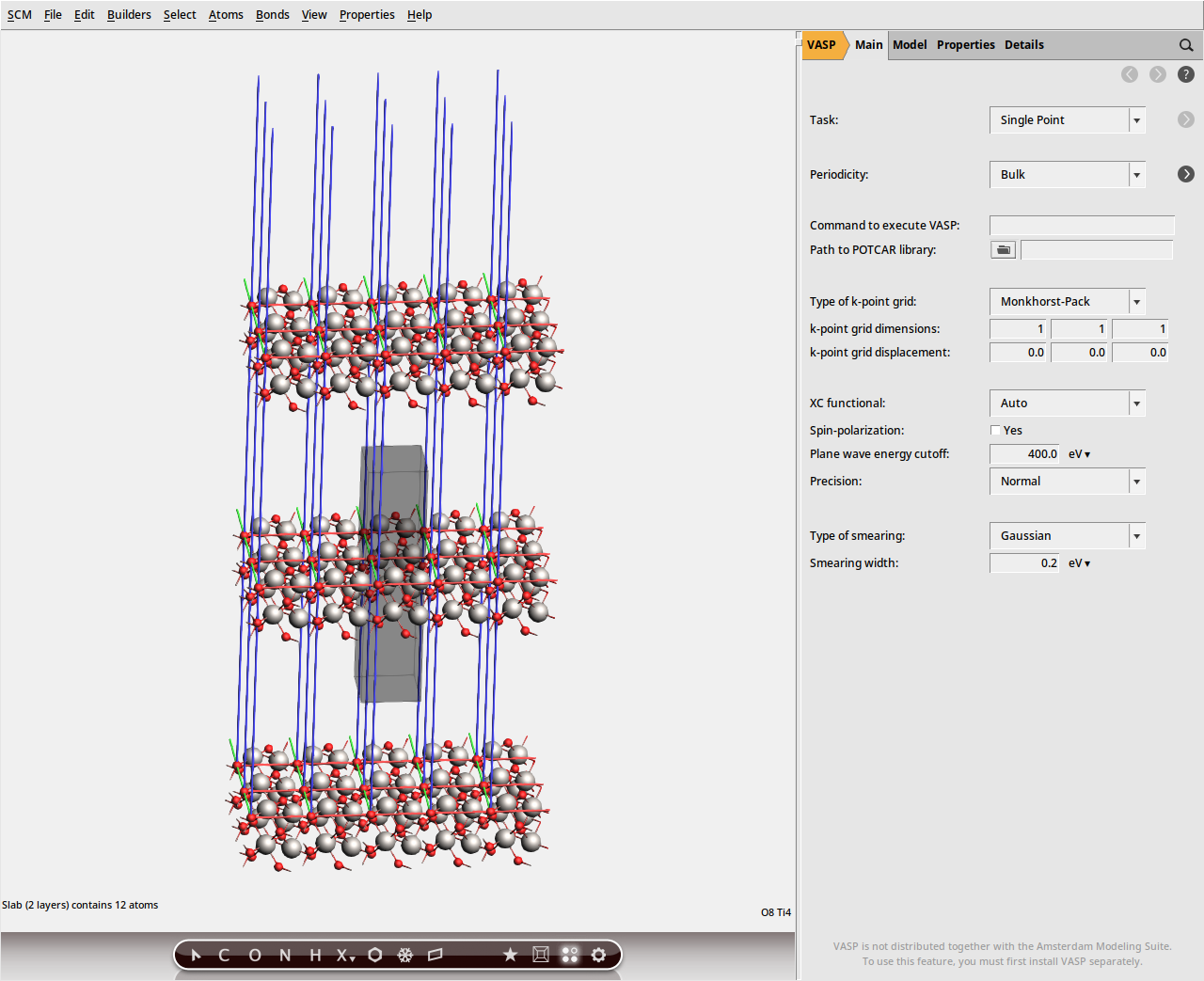
Next, we create a 2-layer (001) slab. (Because the crystal unit cell contains 2 atomic layers, the resulting slab will actually be 4 atomic layers thick).
(0, 0, 1) as the Miller indices.2 (default).This should create a 2-layer slab in the 3D area on the left. Select View → View Direction → Along X-axis from the menu bar, or rotate the system so that the z-axis (shown as a blue line) is roughly displayed vertically. This will give you a side view of the slab.
Note
It may happen that bonds are drawn between adjacent Ti ions. These bonds do not affect the VASP calculation and can safely be ignored.
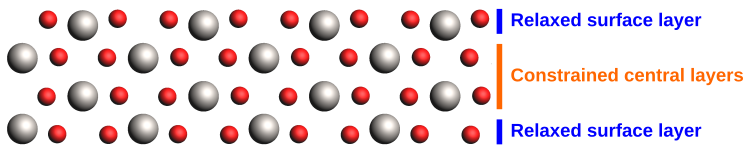
A realistic slab model would typically require additional layers, such that the relaxation of the surface does not affect the bulk structure in the center of the slab. We will instead use geometry constraints here to reduce the size of our model.
All VASP calculations are performed under 3D periodic boundary conditions. Therefore, slabs are necessarily separated by a vacuum gap in the surface normal direction.
 to visualize a few periodically repeated images of the central cell.
to visualize a few periodically repeated images of the central cell.Note that the default vacuum gap is quite large. Because VASP uses a plane wave basis set, a large vacuum gap will increase the computational cost. For this reason, one typically wants to reduce the size of the vacuum gap as much as possible, while still keeping the periodic images from interacting with one another. For this tutorial, we will use a small vacuum gap of around 8 Å.
0.0 0.0 14.0. (Note: this c lattice vector now refers to the slab system including the vacuum gap, and no longer to the original unit cell of the crystal). so that only a single periodic image is displayed.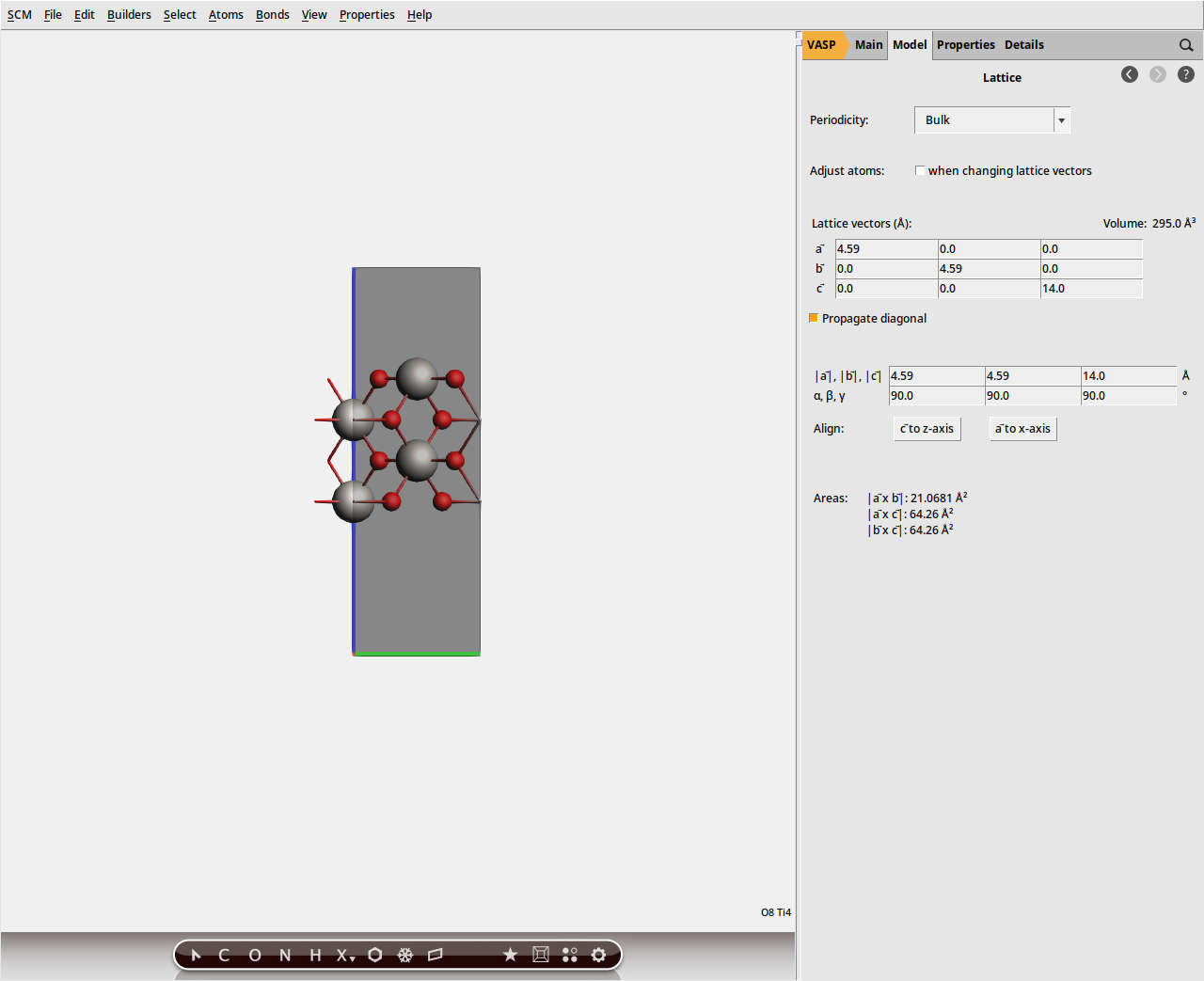
Note
Proper procedure is to systematically vary the vacuum gap for the particular system at hand. Calculated quantities (such as the surface energy, adsorption energies, work function, etc.) should converge as the vacuum gap is increased. One can then select the smallest vacuum gap while still obtaining converged values.
Step 4: Set the VASP settings¶
3 3 1.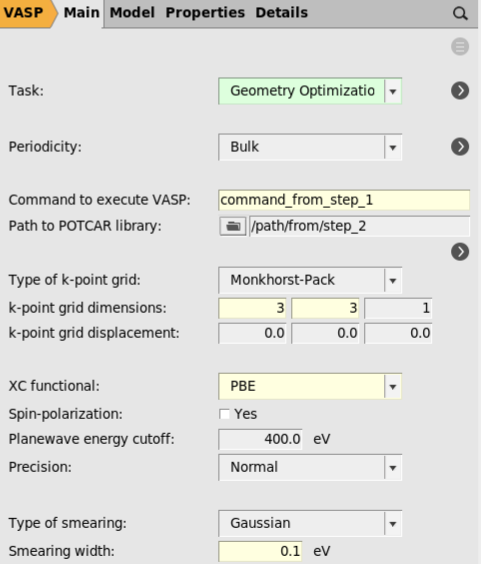
Note
The k-point grid dimensions and plane wave energy cutoff would normally be obtained through convergence tests.
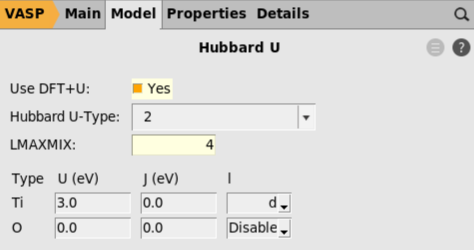
The main VASP input file, INCAR, contains a large number of settings for modifying technical aspects of the calculations. AMSinput does not provide dedicated panels for all of these options. Instead, the user is allowed to add specific settings to the INCAR directly. To illustrate this, we will modify the self-consistent-field algorithm.
ALGO = Fast in the Additional INCAR options text box.Step 5: Set the AMS settings¶
We will use constraints to restrict the geometry relaxation to the surface layers.
 button next to selected atoms (fix position).
button next to selected atoms (fix position).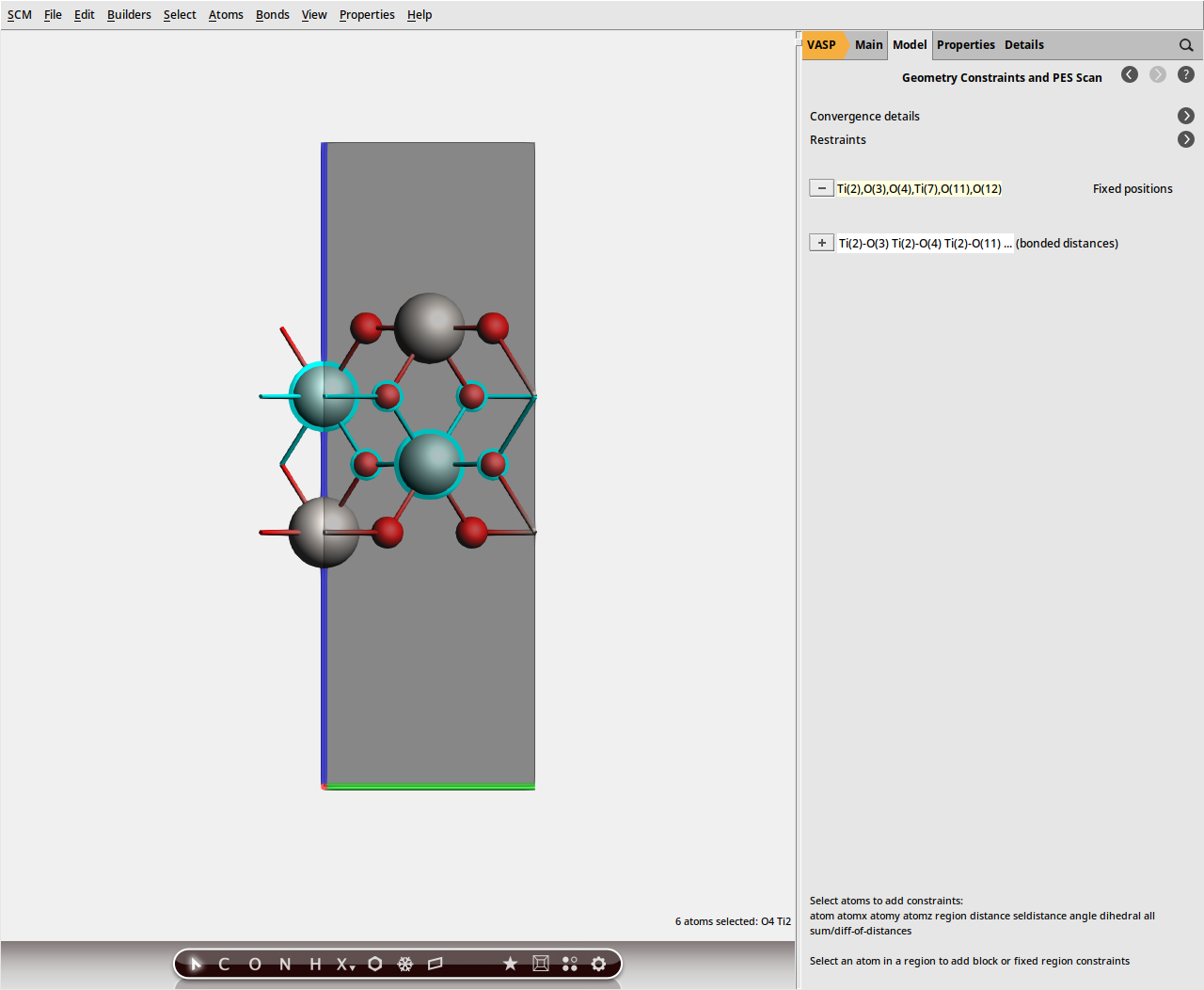
VASP_PBE_U_TiO2_slab.ams.Note
When running VASP via the AMS driver, the geometry optimization settings and constraints are set for the AMS driver and not for VASP. This means that the VASP geometry convergence criteria, such as EDIFFG, do not need to be provided. The Selective Dynamics option will also be handled automatically by AMS.
Step 6: Run the job¶
After the calculation has finished, visualize the geometry optimization in AMSmovie.
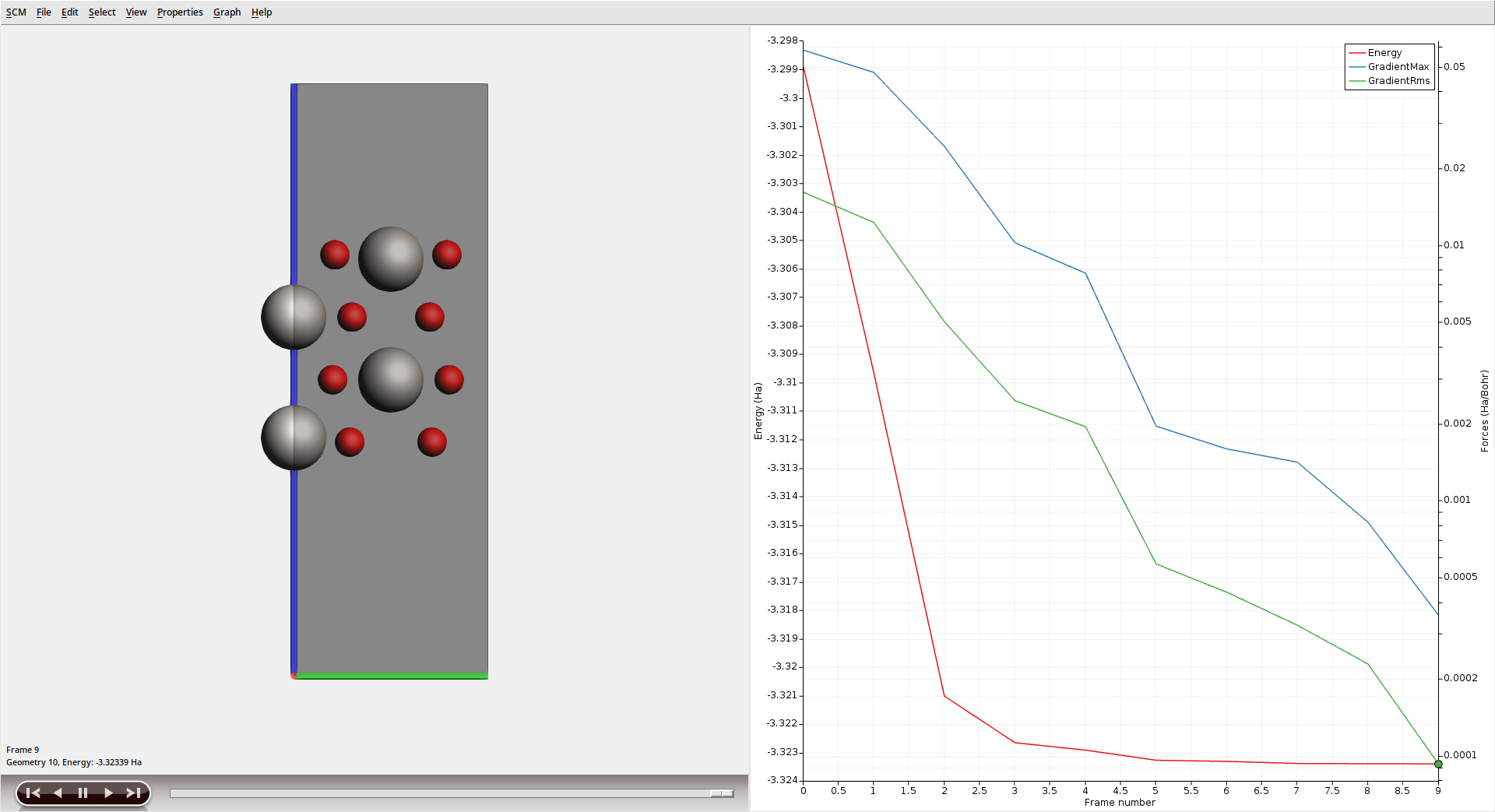
The Ti atoms in the outer layers can be seen to relax inwards towards the bulk as the optimization progresses.